 History: An 8 year old otherwise healthy boy was found to have a left scrotal mass. An ultrasound showed the left testis to be “inhomogeneous†and surrounded by a tumor confined to the scrotum. The right testis was normal. Regional inguinal lymph nodes were not enlarged and an abdominal CT scan was unremarkable.
History: An 8 year old otherwise healthy boy was found to have a left scrotal mass. An ultrasound showed the left testis to be “inhomogeneous†and surrounded by a tumor confined to the scrotum. The right testis was normal. Regional inguinal lymph nodes were not enlarged and an abdominal CT scan was unremarkable.
A left orchiectomy specimen included a 7.0 x 3.5 x 2.5 cm grey-tan solid tumor that surrounded and compressed an uninvolved 1.5 cm testis.
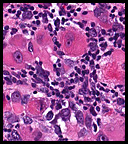
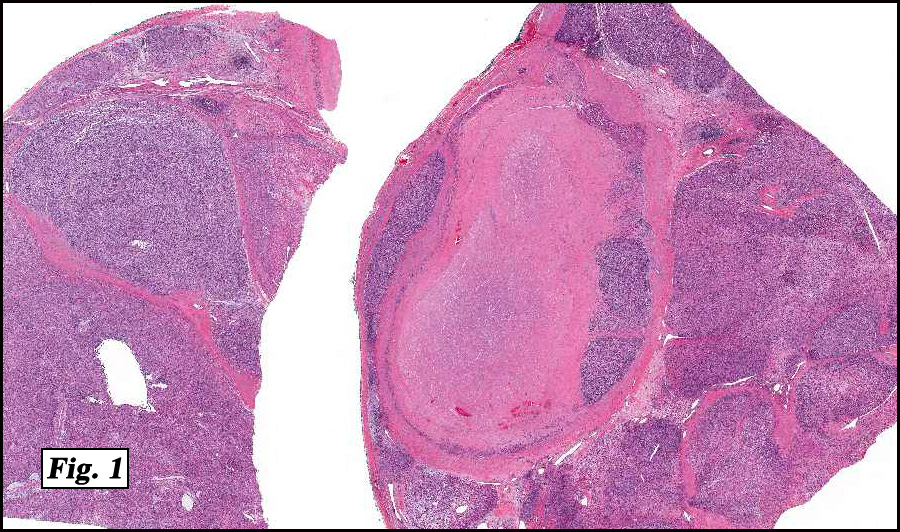
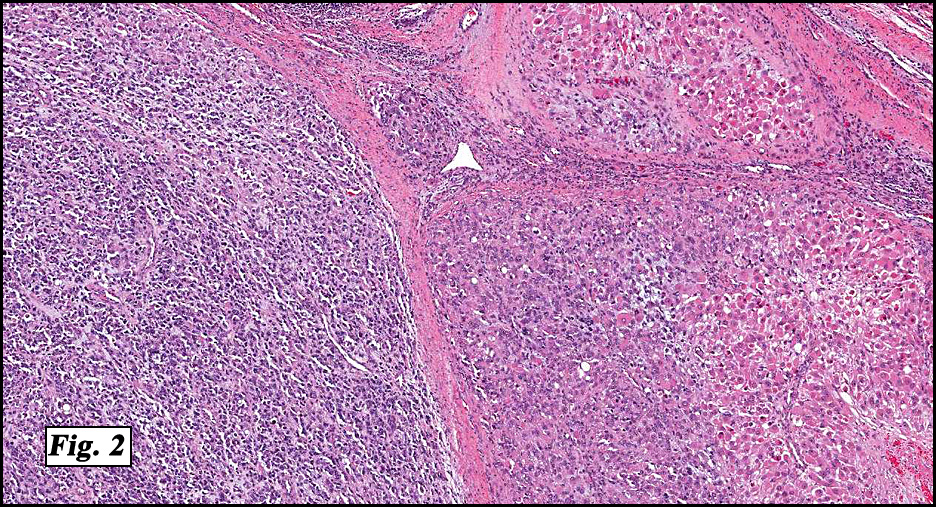
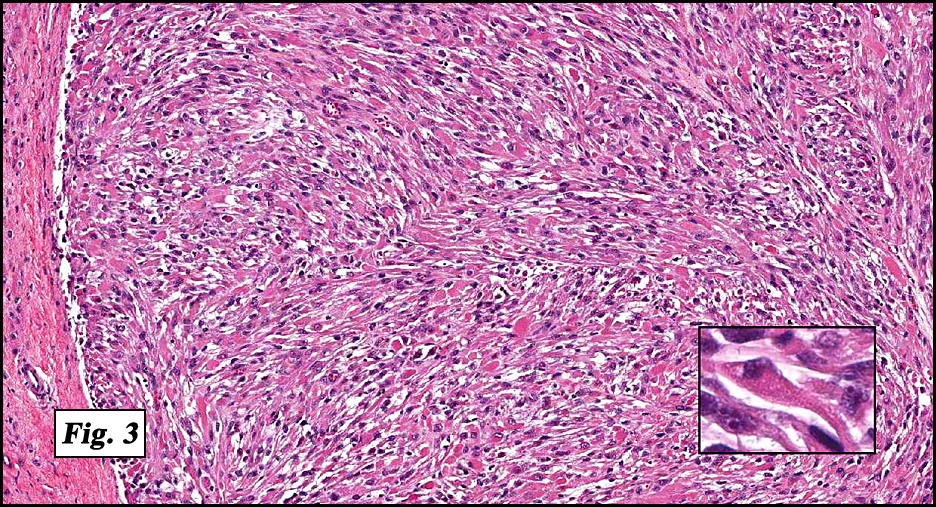
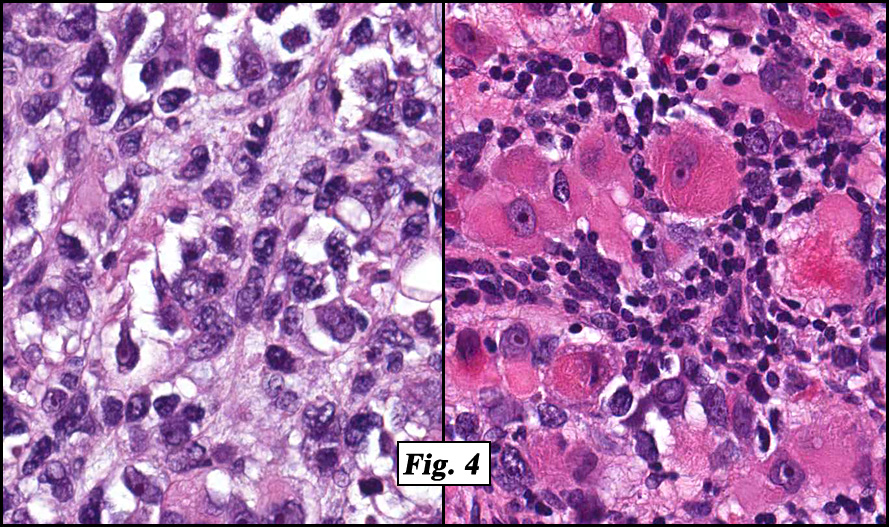
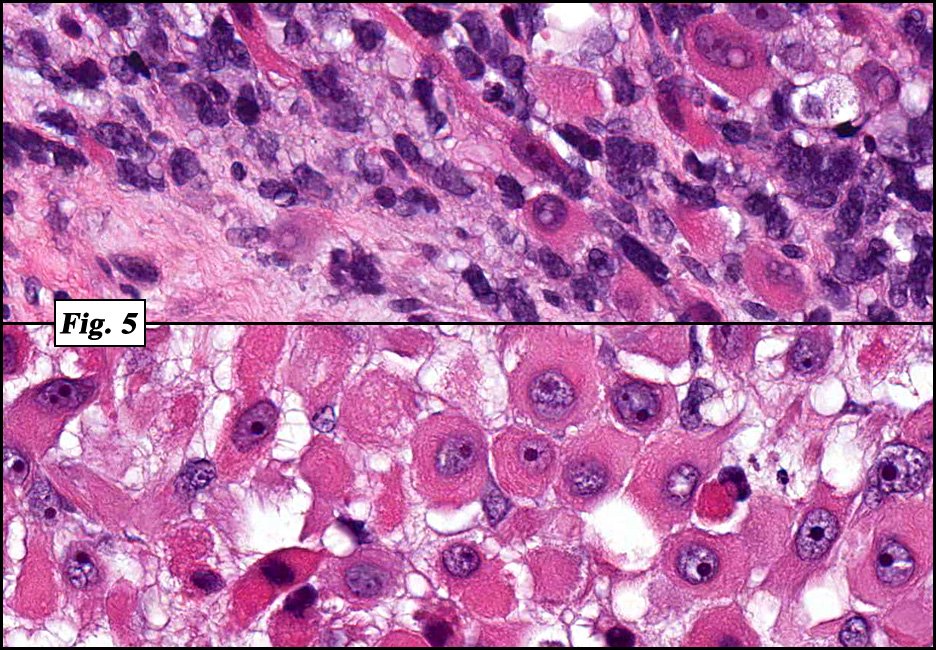
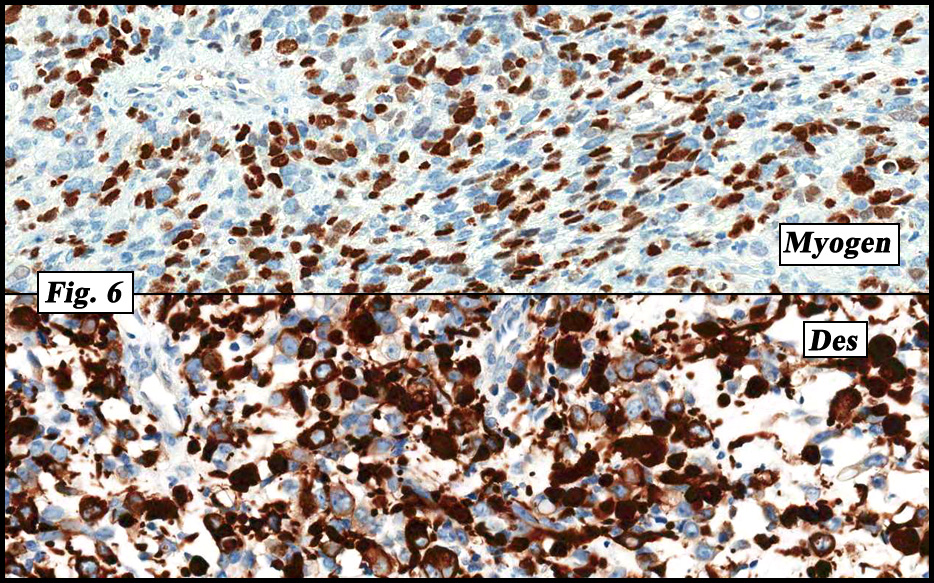
Microscopically the tumor was solid, partly necrotic and highly cellular (Fig. 1). It was partially compartmentalized by fibrous bands separating two populations of cells which varied in amounts of cytoplasm (Fig. 2). Some regions showed spindling with occasional fusiform cells displaying cross striations (Fig. 3). Most of the tumor, however, consisted of areas of primitive small rounded hyperchromatic cells with limited cytoplasm, and other regions having an admixture of primitive cells with larger, more differentiated cells containing abundant eosinophilic cytoplasm and concentric cytoplasmic striations (Fig. 4,Fig. 5). Immunohistochemical stains showed the tumor to have moderate nuclear staining for myogenin (“myogenâ€) and strong cytoplasmic decoration for desmin (“desâ€) (Fig. 6).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Diagnosis: “Paratesticular Embryonal Rhabdomyosarcomaâ€
Krishna Ahuja MD, Donald R. Chase MD, Anwar Raza MD, Craig Zuppan, MD
Department of Pathology, Loma Linda University and Medical Center, Loma Linda, California
California Tumor Tissue Registry, Loma Linda, California
Discussion: Rhabdomyosarcoma (RMS) is a malignant mesenchymal tumor, usually of childhood, showing striated muscle differentiation. It generally arises in body regions with skeletal muscle, though it may also occur in locations lacking these elements such as the prostate or nasopharynx. As a result, RMS is currently felt to devolve not from myocytes, but from primitive mesenchyme having myogenic tendencies. Although the etiology is not known, it is associated with:
Genetic Disorders:
Neurofibromatosis type 1
Adenomatous polyposis coli
Beckwith-Wiedemann syndrome
Gorlin nevoid basal cell carcinoma syndrome
Developmental Disorders:
Congenital lung cyst
Central nervous system malformations
Genitourinary malformations
Fetal alcohol syndrome
Traditionally the morphological classification of RMS includes three main types, each having slightly different ages of occurrence:
• Embryonal rhabdomyosarcoma (infants, children, adolescents, young adults)
• Alveolar rhabdomyosarcoma (adolescents, young adults)
• Pleomorphic rhabdomyosarcoma (adults)
The WHO, however, currently recommends a classification by subtype which emphasizes prognosis.
Prognostic Group and Type % of Cases 5-year Survival
Superior:
Botryoid embryonal RMS 6% 95%
Spindle cell embryonal RMS 3% 88%
Intermediate:
Embryonal RMS, NOS 49% 66%
Poor:
Alveolar RMS 31% 54%
Undifferentiated sarcoma 3% 40%
*(From Newton, 1995 and CTTR seminar of June, 2008 by Cheryl Coffin)
Embryonal RMS
ERMS accounts for approximately 50 % of all RMS mostly affecting children younger than 10 years of age, it also occurs in adolescents and young adults and is uncommon in patients older than 40 years of age. The tumor seems to display various stages of myogenesis and its patterns are variable, ranging from poorly differentiated tumors that are difficult to diagnose without immunohistochemical or electron microscopic examination, to well-differentiated neoplasms that resemble fetal or even adult muscle. The percentage of differentiated rhabdomyoblasts serves as a rough estimate of differentiation. Tumors with mostly primitive small cells tend to do worse than those dominated by larger, rounded rhabdomyoblasts. Two important sub-types with more favorable prognoses are:
Spindle cell RMS: Usually involves paratesticular soft tissue, followed by the head and neck area. Histologically it is composed almost exclusively of elongated fusiform cells with cigar-shaped nuclei and prominent nucleoli. Cross striations are more readily discernible in this subtype than in others.
Botryoid RMS (BRMS): Most commonly found in mucosa-lined hollow organs such as nasal cavity, nasopharynx, bile duct, urinary bladder and vagina. According to ICR criteria, a “cambium layerâ€, characterized by a subepithelial condensation of tumor cells separated from an intact surface epithelium by a zone of loose stroma, must be present to recognize this variant.
Alveolar RMS
This tumor occurs more often in limb girdles than does BRMS and usually arises in adolescents. The classic alveolar pattern consists of anastomosing fibrovascular septa lined by discohesive tumor sometimes accompanied by multinucleated cells. The neoplasm is aggressive and may present with distant metastasis. Curiously, in addition to lung, it may metastasize to breast or the bone marrow where it can cause a leukemic-like presentation.
Pleomorphic RMS
Accounting for no more than 10% of the tumors, the “adult form†of RMS is rare. It usually arises in the limbs and appears to be less chemosensitive than the other variants. The histology is that of a pleomorphic sarcoma (nos) showing positivity for muscle markers such as myogenin, desmin, etc. The expected 5-year survival is only around 20%. Of interest is that it may be associated with prior radiation to or near the primary site.
With combination therapy, especially improved chemotherapy, the prognosis of RMS has greatly improved through the years. Despite the dramatic improvement in treatment of RMS, early diagnosis and accurate classification remains vital. Continued therapeutic success is expected as we enter the molecular era with the potential for targeted therapies.
Suggested Reading:
Stout AP. Rhabdomyosarcoma of the skeletal muscles. Ann Surg 123:447, 1946.
Newton WA Jr, et al. Classification of rhabdomyosarcomas and related sarcomas. Pathologic aspects and proposal for a new classification – an Intergroup Rhabdomyosarcoma Study. Cancer 76:1073, 1995.
Parham DM: Pediatric Neoplasia: Morphology and Biology, Chapter 5, pp 87-100, Lippincoot-Raven Publishers, Philadelphia 1996.
Cheryl M. Coffin: “The New International Rhabdomyosarcoma Classification, Its Progenitors, and Considerations beyond Morphology.†Advan Anat Pathol, Vol 4, No 1, pp1-16, 1997.
Enzinger and Weiss’: Soft Tissue Tumors 5th Ed. Editors SW Weiss, JB Goldblum. Mosby, Inc. pp 595-632, 2008.