 History: A 14-year-old Caucasian girl presenting with worsening abdominal pain was found to have marked splenomegaly and moderate pancytopenia. She was admitted with the clinical impression of acute leukemia. A bone marrow biopsy was performed. Her past medical history was remarkable only for a hip fracture after falling from the bicycle about 4 years previously (Fig. 1).
History: A 14-year-old Caucasian girl presenting with worsening abdominal pain was found to have marked splenomegaly and moderate pancytopenia. She was admitted with the clinical impression of acute leukemia. A bone marrow biopsy was performed. Her past medical history was remarkable only for a hip fracture after falling from the bicycle about 4 years previously (Fig. 1).
{kind=link}
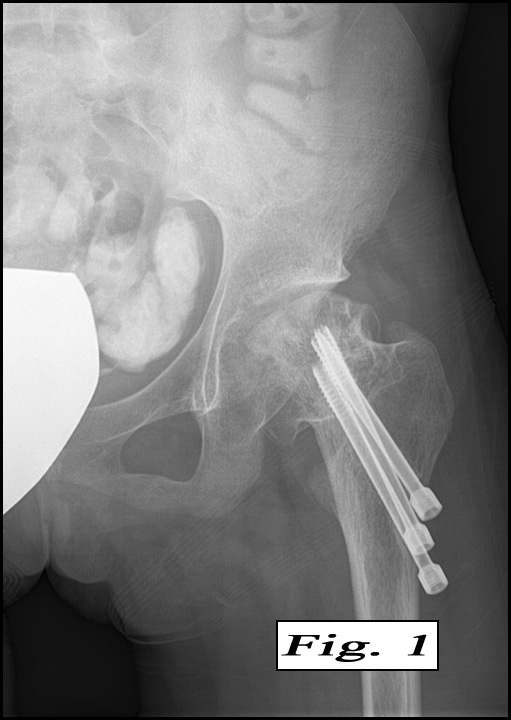
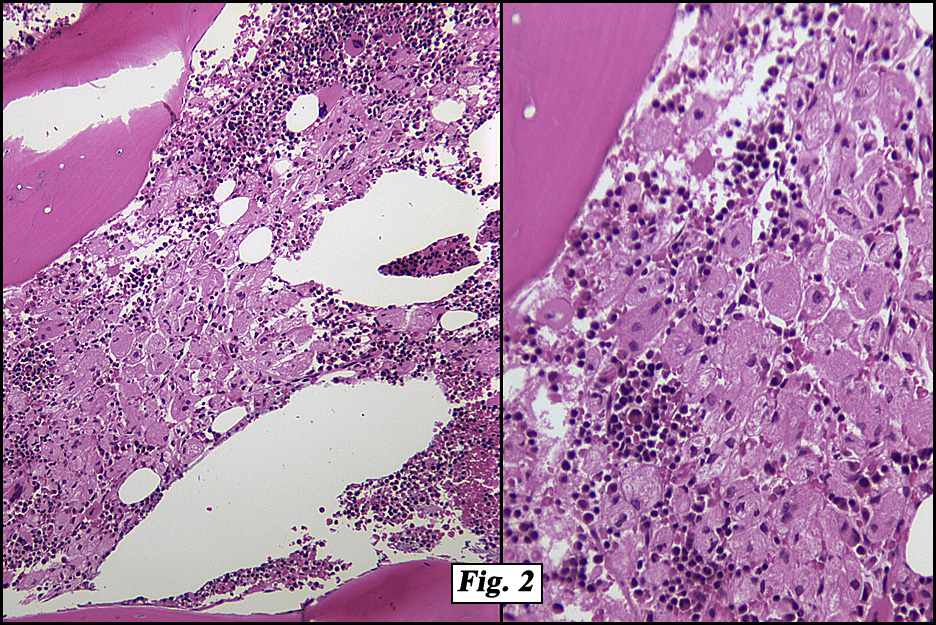
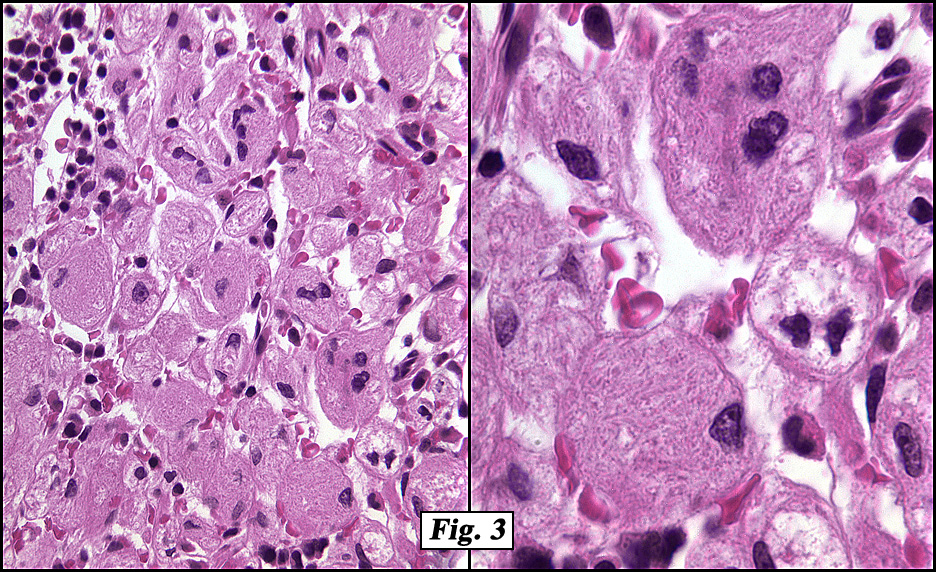
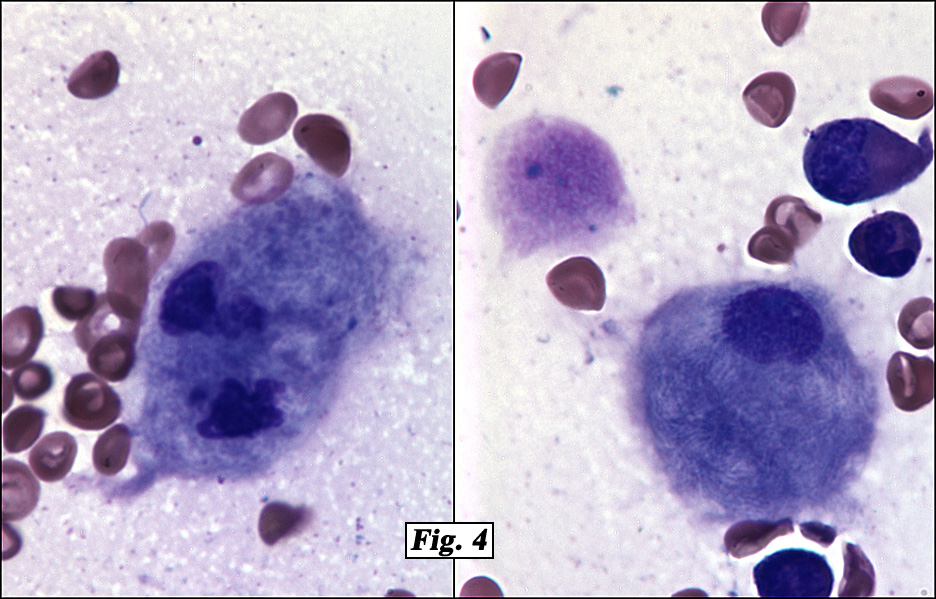
The peripheral blood showed marked anisopoikilocytosis, mild microcytosis, hypochromia and moderate thrombocytopenia. A bone marrow aspirate and biopsy (Fig. 2) was normocellular (~85%, cellularity) with the cellular composition reflecting active trilineage hematopoiesis with orderly complete maturation. Individual as well as numerous clusters and aggregates of large cells were present with abundant granular or fibrillary, blue-gray cytoplasm with a “wrinkled tissue paper-like” appearance with abundant lightly PAS-positive fibrillary material in the cytoplasm (Figs. 3,4), and eccentric nuclei. A CD68 immunohistochemical stain was positive as was an iron stain.
{kind=link}
{kind=link}
{kind=link}
Flow cytometry showed slightly increased numbers of hematogones (approximately 8% total events) with no evidence of aberrant antigen expression or of a myeloid or lymphoid neoplasm. Cytogenetics showed a normal female karyotype, 46,XX. An enzyme panel demonstrated normal activities for all lysosomal enzymes with the exception of beta glucosidase (glucocerebrosidase), which was low. The combined findings were supportive of a diagnosis of Gaucher disease (GD).
Diagnosis: “Gaucher Diseaseâ€
Mingyi Chen MD, Donald R. Chase MD, Jun Wang MD,
Antranik Bedros MD
Department of Pathology and Human Anatomy,
Loma Linda University and Medical Center, Loma Linda, CA
California Tumor Tissue Registry, Loma Linda, CA
Discussion: Gaucher disease was first described by Philippe Gaucher in his doctoral thesis in 1882, when he hypothesized that infiltration of enlarged cells in a spleen represented a “neoplasmâ€. The biochemical basis for the disease was elucidated 83 years later (1965) by Dr. Roscoe Brady of the NIH.
GD is the most common of the lysosomal storage diseases and is inherited in an autosomal recessive fashion. It is caused by a deficiency of the enzyme glucocerebrosidase, leading to an accumulation of its substrate glucocerebroside in the mononuclear phagocyte system, especially Kupffer cells in the liver, osteoclasts in bone, microglia in the central nervous system, alveolar macrophages in the lungs, and histiocytes in the spleen, lymph nodes, bone marrow, gastrointestinal and genitourinary tracts, as well as the peritoneum.
Clinical signs and symptoms include painless hepatosplenomegaly, hypersplenism leading to anemia, neutropenia and thrombocytopenia (with an increased risk of infection and bleeding) and rare cirrhosis of the liver.
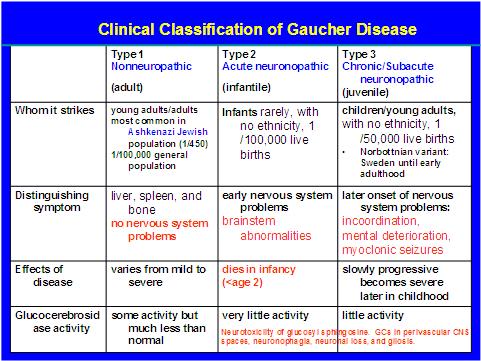
The disease is usually classified into three types (Table 1). Neurological symptoms such as convulsions, dementia, ocular muscle apraxia, mental retardation, and myoclonus can be seen as well as osteoporosis, hypertonia, apnea, Erlenmeyer flask deformity of the distal femur and a yellowish-brown skin pigmentation. Noticeably absent are demonstrable cardiac, renal or pulmonary symptoms.
Gaucher disease is most common in the Ashkenazi Jewish population. In a population survey involving 2000 Ashkenazi Jews, the gene frequency was 0.034. Approximately 6.8% of the Jewish population is heterozygous for GD, and, based on this, the expected birth frequency is 1 in 1000. The disease is also relatively common among the Norbottnian population in Northern Sweden.
Genetic studies have shown that the glucocerebrosidase gene (GBA) is located on chromosome 1q21 and over 200 mutations of this gene have been reported as being associated with GD. In the Ashkenazi Jewish population, the predominant point mutation is N370S. This mutation accounts for ~75% of the mutant alleles in Jewish patients and ~30% in non-Jewish patients. It predisposes to type 1 disease and precludes neurological involvement.
A frame shift mutation of 84GG is relatively common in the Jewish population. The L444P mutation is common in the Norbottnian population and the homozygous state has a very high association with the neuronopathic variants of GD. Recent genetic analysis suggested that the N370S and 84GG mutations each originated in a single founder/progenitor of/in the Ashkenazi Jewish population. Thus, the founder effect followed by genetic drift rather than an evolutionary advantage for heterozygotes best explains the high frequency of these mutations in Ashkenazi Jews.
It has been suggested that despite considerable uncertainty about the demographic history of Ashkenazi Jews and their ancestors, the available genetic data is consistent with a founder effect resulting from a severe bottleneck in population size between 1100 A.D. and 1400 A.D. and an earlier bottleneck in 75 A.D., at the beginning of the Jewish Diaspora. A founder effect could account for the relatively high frequency of alleles causing the lysosomal storage disorders, including Tay-Sachs disease and Gaucher disease, if the disease-associated alleles are recessive in their effects on reproductive fitness.
But the diagnosis is usually based on identification of Gaucher cells in a bone marrow aspirate. However, enzymatic assay to evaluate beta-glucocerebrosidase activity in leukocytes combined with molecular analysis is now considered to be the gold standard. Interestingly, targeted mutational analysis can be used to aid diagnosis in the Ashkenazi Jew population, where four specific sites (N370S, L444P, 84GG, IVS2+1) account for approximately 90% of the disease-causing alleles. Sequence analysis of the GBA coding region can be used to detect mutations in affected individuals in the general population. DNA testing therefore currently provides the most reliable means of identifying carriers, and carrier testing is recommended for all close relatives of a confirmed GD patient.
The differential diagnosis of Gaucher’s disease is that of other lipid accumulation abnormalities including:
• Pseudo-Gaucher’s disease in which Gaucher-like cells can be found in hematologic abnormalities such as CML, AML, CLL, Hodgkin lymphoma, multiple myeloma, and ITP. Although the cells cannot be distinguished from true Gaucher cells on H&E stained sections, iron staining will generally be negative (but positive in true Gaucher cells).
• Niemann-Pick disease is caused by a deficiency in sphingomyelinase which leads to accumulation of sphingomyelin in the cytoplasm of macrophages. The globules are small and relatively uniform in size, sometimes described as mulberry-like in appearance. Foamy macrophages similar to Niemann-Pick cells may be seen in any disorder that is associated with massive cell destruction that overloads the capacity of the cellular machinery to digest lipids (e.g., thalassemias, sickle cell anemia, ITP, chronic renal failure). The vacuoles may be larger and more irregular in size than those seen in true Niemann-Pick cells. As compared to Gaucher cells, the cytoplasm in these macrophages is foamy and vacuolated as opposed to fibrillary.
• Sea blue histiocytes may be found in small numbers in any disorder associated with massively increased intramedullary cell destruction, such as chronic myelogenous leukemia. The macrophage cytoplasm is filled with insoluble lipid pigment, called ceroid, which is thought to represent partially digested globosides derived from cell membranes. As compared to Gaucher cells, these cells stain more intensely blue with Wright-Giemsa and the inclusions are globular rather than fibrillary.
• In Tay Sachs disease (hemoaminidase A deficiency), there is accumulation of GM2 ganglioside in the heart, liver and spleen. Involvement of the central nervous system with vacuolated neurons is predominant. Patients present at 6 months of age and the disease is fatal by age 2-3 years of age.
• Pompe disease (acid maltase deficiency) is characterized by glycogen accumulation in hepatocytes and muscle cells, but the primary pathologic derangement is in skeletal and cardiac muscle, not bone marrow.
Enzyme replacement therapy with a recombinant glucocerebrosidase known as imiglucerase (Cerezyme) is the mainstay of treatment for GD which became the first successfully managed lipid storage disease. Hepatosplenomegaly, anemia, and thrombocytopenia usually improve within 6 months. Unfortunately, there is no effect on neurological symptoms because it does not cross the blood-brain barrier. Future treatments may include gene therapy and intervention with chemical chaperones.
Suggested Reading:
1. Gaucher PCE. De l’epithelioma primitif de la rate, hypertrophie idiopathique de la rate sans leucemie. Academic thesis, Paris, France, 1882.
2. Brady RO, Kanfer JN, Shapiro D.”Metabolism of glucocerebrosides. II. Evidence of an enzymatic deficiency in Gaucher’s disease”. Biochem. Biophys. Res. Commun 18:221-5. PMID 14282020, 1965.
3. Jmoudiak M, Futerman AH. Gaucher disease: pathological mechanisms and modern management. BJH 129:178-188, 2005.
4. Beutler E, Nguyen NJ, Henneberger MW, et al. Gaucher disease: gene frequencies in the Ashkenazi Jewish population. Am J Hum Genet 52:85-88, 1993.
5. Cytoplasmic inclusions in bone marrow cells. In: Color Atlas of Hematology. Glassy E, ed. Northfield, IL: CAP 320-327. 1998.
6. Elstein D, Abrahamov A, Hadas-Halpern I, Zimran A. Gaucher’s disease. Lancet 358:324-327, 2001.
7. Zimran A, Altarescu G, Rudensky B, Abrahamov A, Elstein D. Survey of hematological aspects of Gaucher disease. Hematology 10:151-156, 2005.
8. Shiran A, Brenner B, Laor A, Tatarsky I. Increased risk of cancer in patients with Gaucher disease. Cancer 72:219-224, 1993.
9. Aerts JMFG, Hollak CEM, van Breeman M, Maas M, Groener JEM, Boot RG. Identification and use of biomarkers in Gaucher disease and other lysosomal storage diseases. Acta Paediatrica 94:43-46, 2005.
10. Brady RO. Enzyme replacement for lysosomal diseases. Annu Rev Med 57:283-296, 2006.