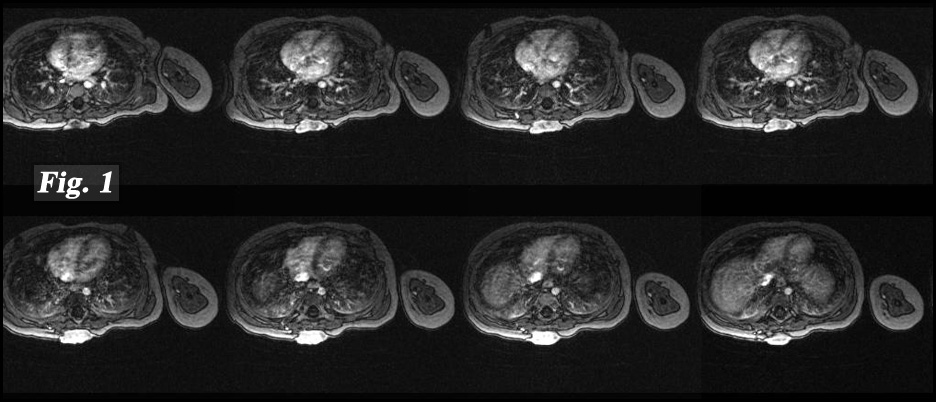
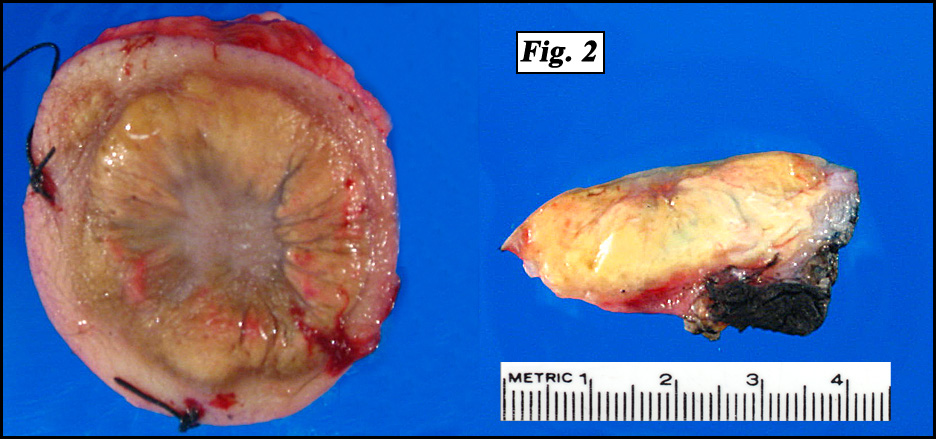
 History: A 16 month-old girl presented with a round nodular mass in her mid upper back that had been present for 12 months. It was found to involve the cutaneous and subcutaneous tissues as well as underlying fascia of the right paraspinal region. It did not infiltrate either bone or muscle (Fig. 1). Excision revealed a 3.0 x 2.5 x 1.0 cm poorly-circumscribed annular, lobulated yellow tumor (Fig. 2).
History: A 16 month-old girl presented with a round nodular mass in her mid upper back that had been present for 12 months. It was found to involve the cutaneous and subcutaneous tissues as well as underlying fascia of the right paraspinal region. It did not infiltrate either bone or muscle (Fig. 1). Excision revealed a 3.0 x 2.5 x 1.0 cm poorly-circumscribed annular, lobulated yellow tumor (Fig. 2).
{kind=link}
{kind=link}
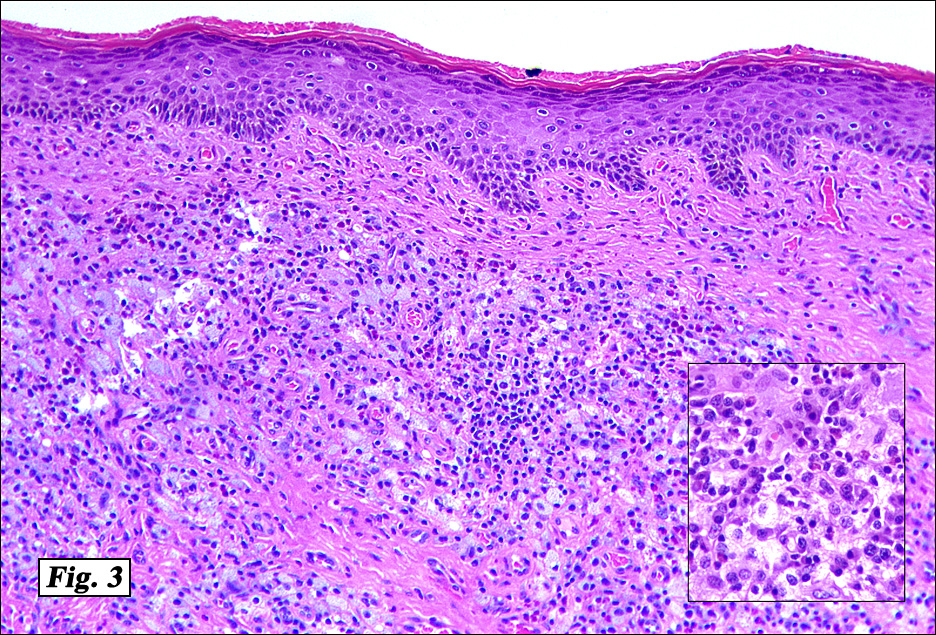
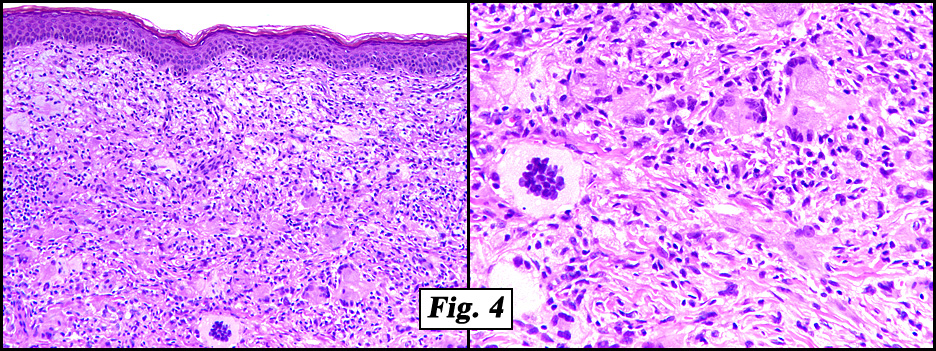
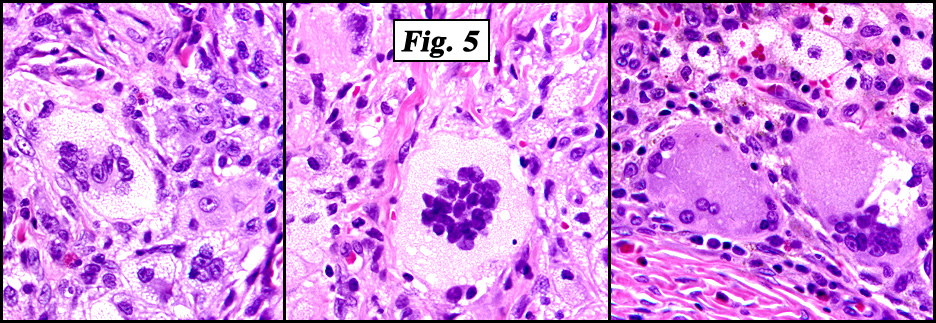
Microscopic examination showed a nodular proliferation of xanthomatous-appearing cells with abundant foamy cytoplasm having spindled cells present at the tumors base (Fig. 3). An admixture of inflammatory cells consisting predominantly of lymphocytes, plasmacytoid monocytes and eosinophils were seen accompanied by scattered Touton giant cells with a wreath-like arrangement of nuclei (Fig. 4,Fig. 5). No cellular atypism was encountered.
{kind=link}
{kind=link}
{kind=link}
Diagnosis: “Juvenile Xanthogranuloma, Skin of Upper Backâ€
Mingyi Chen MD, Donald R. Chase, MD
Department of Pathology, Loma Linda University and Medical Center
California Tumor Tissue Registry
Discussion: Juvenile xanthogranulomas (JXGs) are benign, usually asymptomatic, red-to-yellow papules and nodules that occur in the skin, eyes, and viscera. They generally occur in infancy or childhood and are composed of histiocytic-appearing cells. Approximately 35% of JXGs are found at birth and 71% of cases are reported in the first year of life, but the overall mean presentation age is 22 months. Despite the term “juvenileâ€, 10% manifest in adulthood. The most frequent site is the head and neck, followed by the trunk and upper extremities however JXG has been described anywhere on the skin.
JXG occurs mostly in whites approximately 10 times more frequently than in African Americans. In childhood, JXG occurs predominately in males (1.4:1). Café au lait macules occur in approximately 20% of patients with papular JXG. Niemann-Pick disease, Urticaria pigmentosa, and Neurofibromatosis type 1 (NF1) have been reported to be associated with JXG. Juvenile chronic myelomonocytic leukemia (JMML) has been observed in association with multiple JXGs, and the prevalence is especially high in patients with coexistent neurofibromatosis.
JXG is a tumor described just over 100 years ago:
• In 1905, Adamson first reported JXG in the English literature. He presented a child who developed numerous yellow-white papules on the body in the first 2 weeks of life. He named the entity congenital xanthoma multiplex.
• In 1912, McDonagh renamed the condition nevoxanthoendothelioma (although the condition is now not known to be associated with either nevi or endothelial cells).
• In 1937, Laurb and Lain first reported JXG with visceral involvement.
• In 1949, Blank et al first described ocular involvement.
• In 1954, Helwig and Hackney again re-termed it juvenile xanthogranuloma, reflecting its histopathologic appearance.
The etiology of JXG is not known. The red-to-yellowish papules and nodules of JXG represent collections of differentiated non–Langerhans cell histiocytes. JXG may be a granulomatous reaction of histiocytes to an unidentified stimulus, possibly of either physical or infectious etiology. The appearance of giant cells and foamy lipid-laden histiocytes generally occurs late and apparently is a secondary event, possibly in response to cytokine production by histiocytes. Serum lipid levels are normal and remain normal.
Histological examination of JXG demonstrates a variety of findings. A time-dependent progression exists in the development of the characteristic morphologic features of JXG, which correlates with the age of the lesion. Early biopsy specimens reveal a dense monomorphous histiocytic infiltrate in the dermis. Extension into subcutaneous tissue, fascia, and muscle occurs in approximately one third of cases. Older lesions contain foam cells, Touton giant cells, and foreign body giant cells. A mixed cellular infiltrate of neutrophils, lymphocytes, eosinophils, and (rarely) mast cells may be noted. Old lesions demonstrate fibrosis. The histiocytes contain pleomorphic nuclei, with few or absent mitotic figures, and irregular dense bodies.
The use of special stains is important to differentiate JXG from Langerhans cell histiocytoses (LCH) and other non–Langerhans cell histiocytoses. LCH is characterized by immunohistochemical reactivity for S-100 protein and CD1a surface antigen, generally negative for CD68, HAM56, and Factor Xllla and the presence of Birbeck granules on the electron microscopy. In JXG, histiocytes are positive to antibodies against Factor XIIIa, HAM56, HHF35, KP1 (CD68), Ki-M1P, and Vimentin, and are generally negative to CD1a and S-100. CD4 and CD45 (LCA) may be used to distinguish JXG from dermatofibromas when the latter is heavily lipidized or the former is not. There are several other processes that are contained in the differential diagnosis of juvenile xanthogranuloma. Xanthoma disseminatum is also a non-X histiocytosis, but lacks foreign body-type giant cells and eosinophils both of which are usually present in typical juvenile xanthogranuloma. Papular xanthomas contain foam cells and Touton giant cells, but lack the mixed inflammatory infiltrate found in juvenile xanthogranulomas.
Simple tumor excision usually is curative for JXG except in the very rare systemic types, in which multimodal chemotherapy is indicated.
Suggested Reading:
Enzinger and Weiss: Soft Tissue Tumors (4th ed., Editors Weiss SW & Goldblum J), Mosby, pp 590-597, 2001.
Fletcher CDM, Unni KK, and Mertens F (eds.). World Health Organization Classification of Tumours. Pathology and Genetics of Tumours of Soft Tissue and Bone. IARCPress: Lyon 2002.
Sangueza O, et al. Juvenile Xanthogranuloma: A Clinical, Histopathologic, and Immunohistochemical Study. J Cut Pathol 1995: 22: 327-335
Tahan S, et al. Juvenile Xanthogranuloma. Arch Pathol Lab Med: p 1061, 1989.
Gutmann DH, Gurney JG, and Shannon KM. Juvenile xanthogranuloma, neurofibromatosis 1, and juvenile chronic myeloid leukemia. Arch Dermatol 132(11):1390-1, 1996.