History: A 69-year-old man was hospitalized for 10 days with complaints of easy fatigability. He was found to have dry skin with hyperpigmentation over the lower legs and some cracking of the skin of the lower abdomen. Lab studies showed a sodium level of 129 mEq and a potassium level of 4.5 mEq. The work up discovered chronic active hepatitis, arteriosclerotic heart disease, chronic renal disease, and early COPD. He had a femoral arteriogram to evaluate lower extremity vascular insufficiency, but died 2 months later of apparent cardiac arrest.
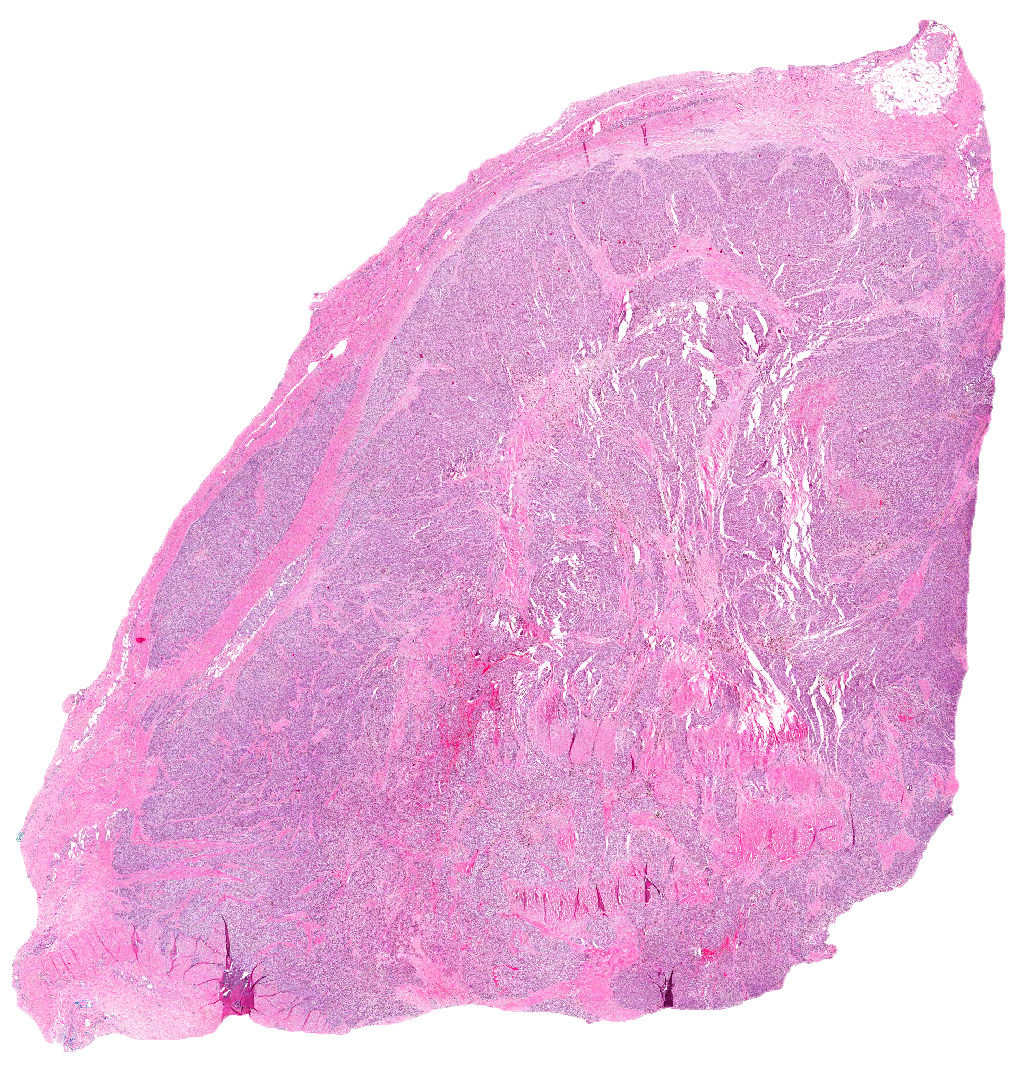
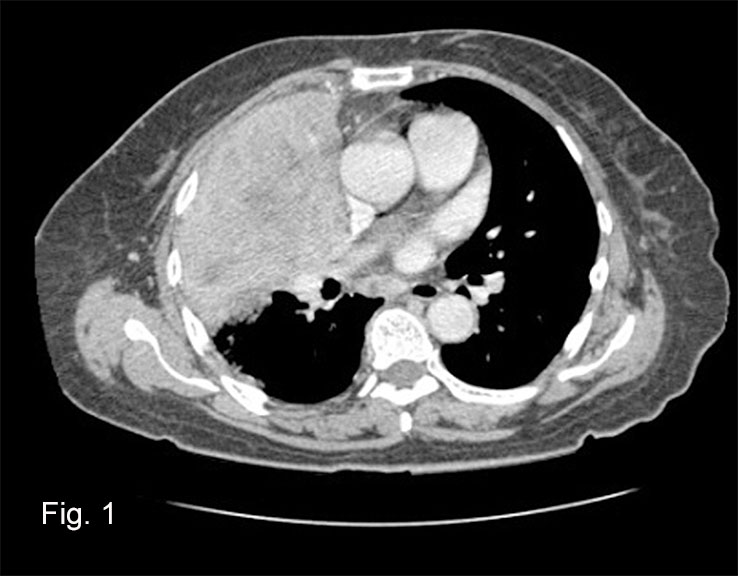
At autopsy, both adrenal glands were enlarged to 5 cm and appeared completely replaced by tumor. Each weighed 30 grams. There was no infiltration of surrounding tissue. Metastases were not seen.
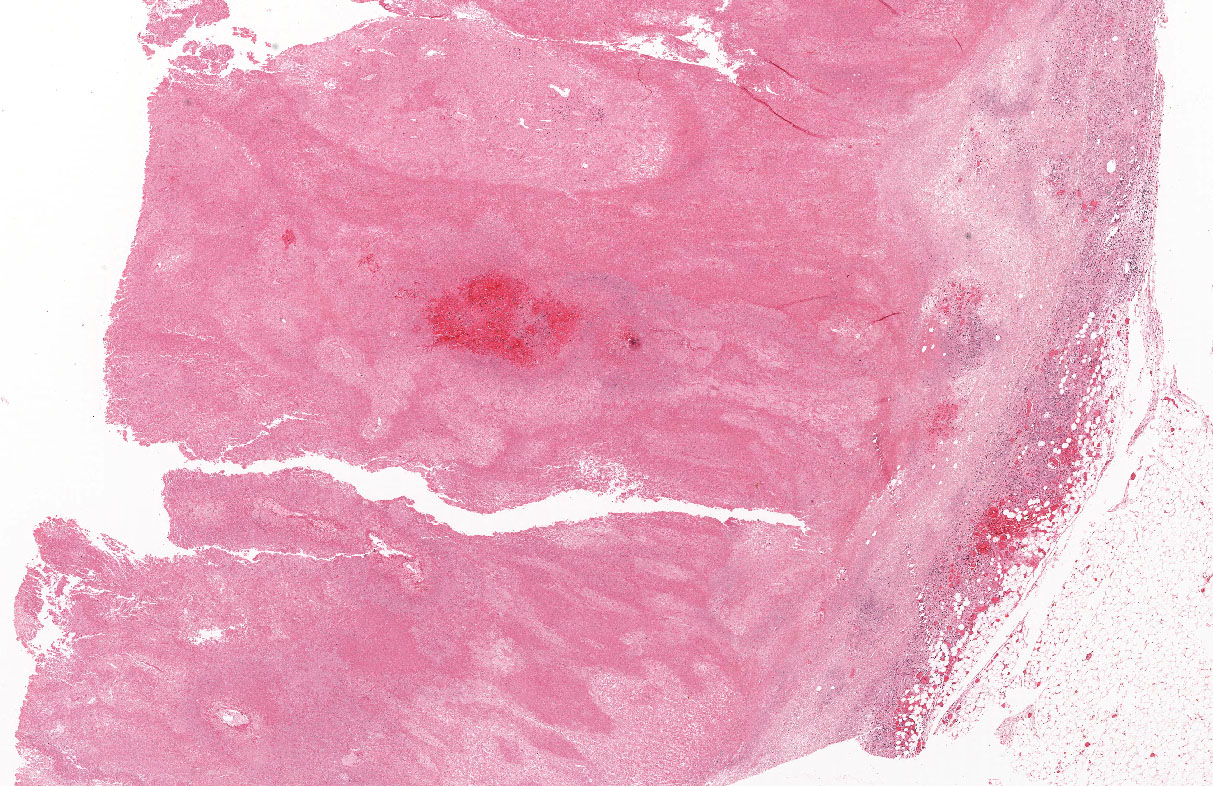
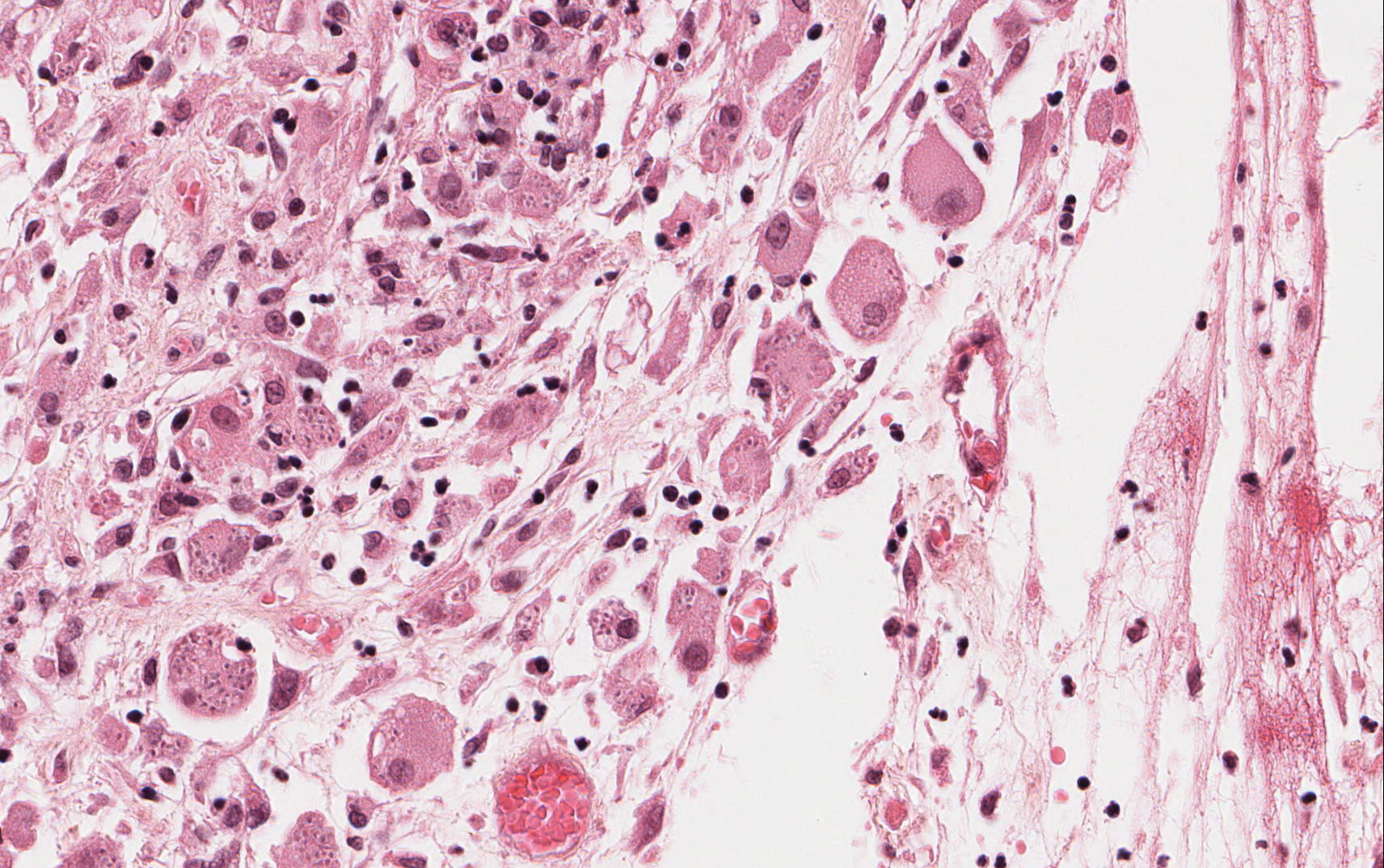
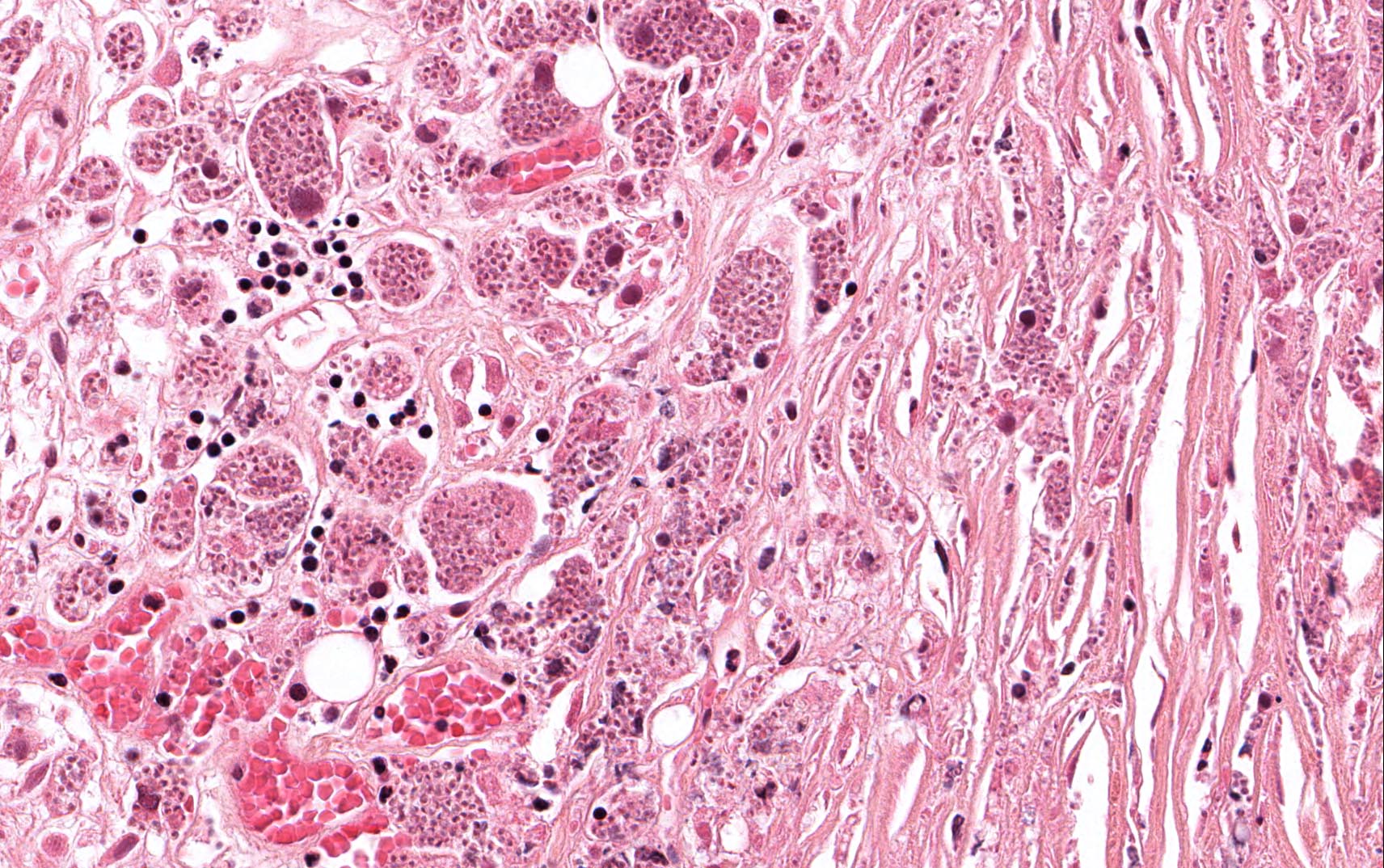
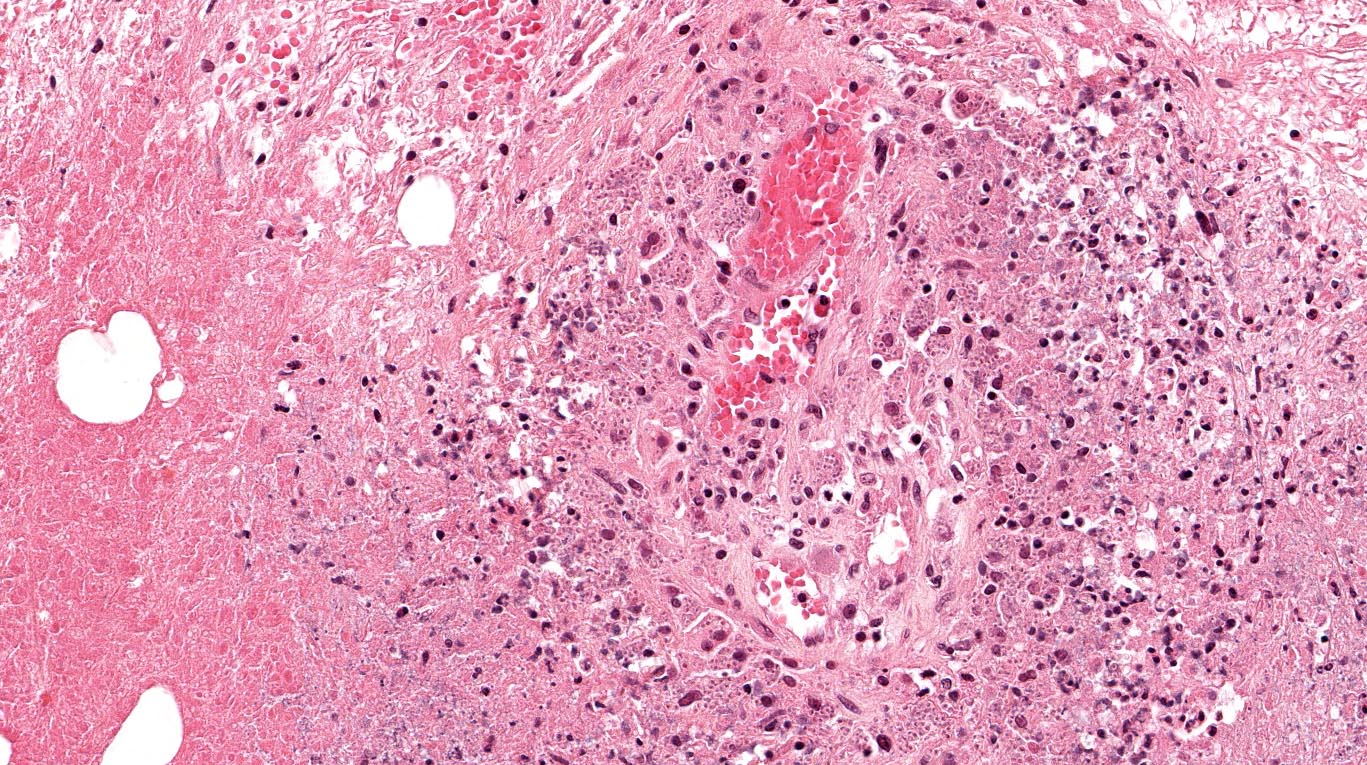
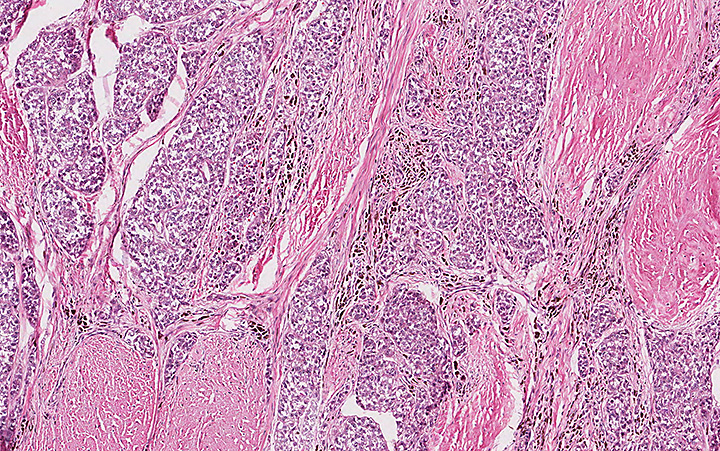
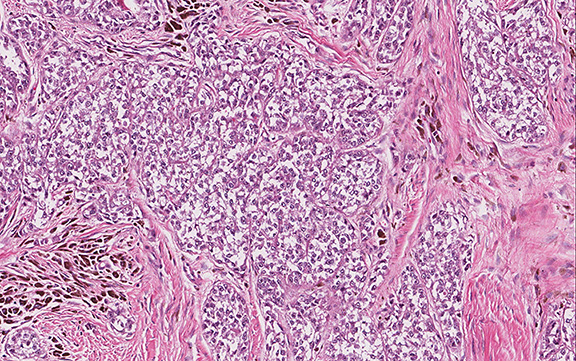
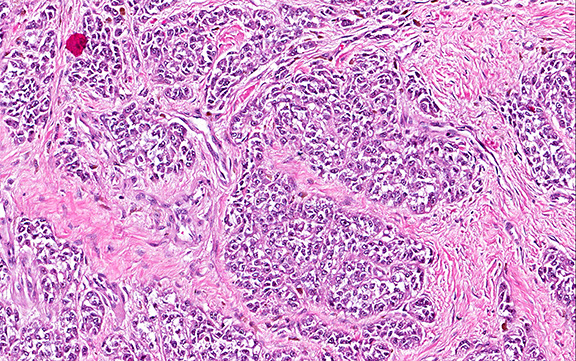
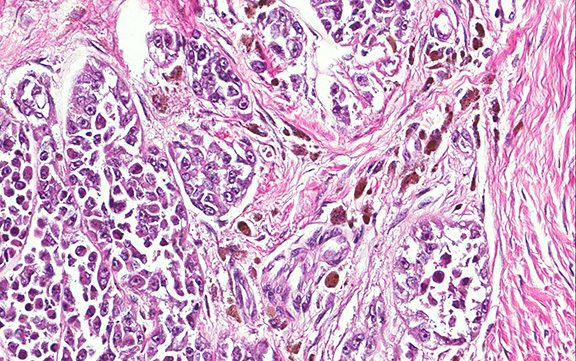
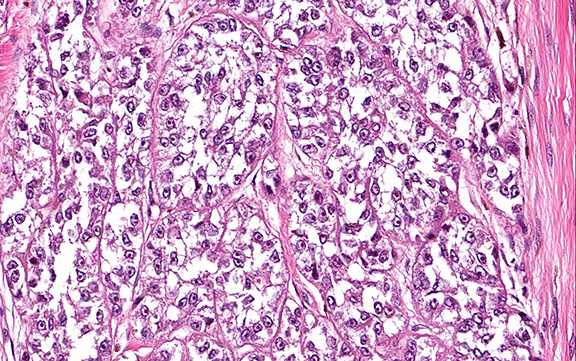
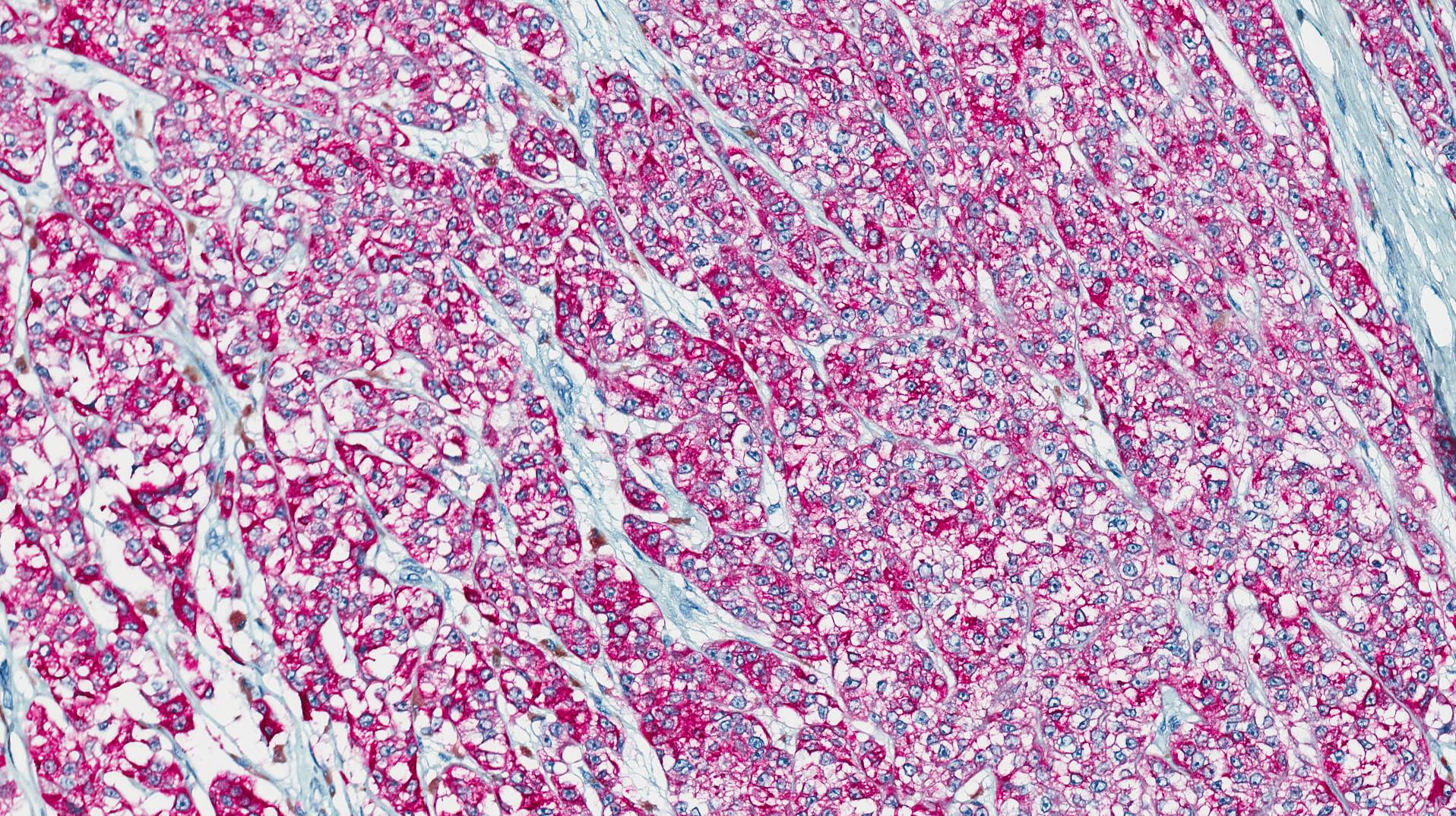
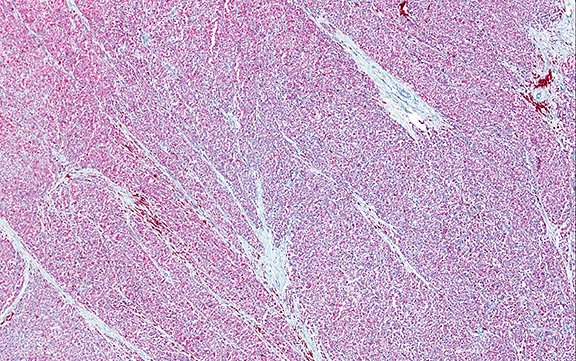
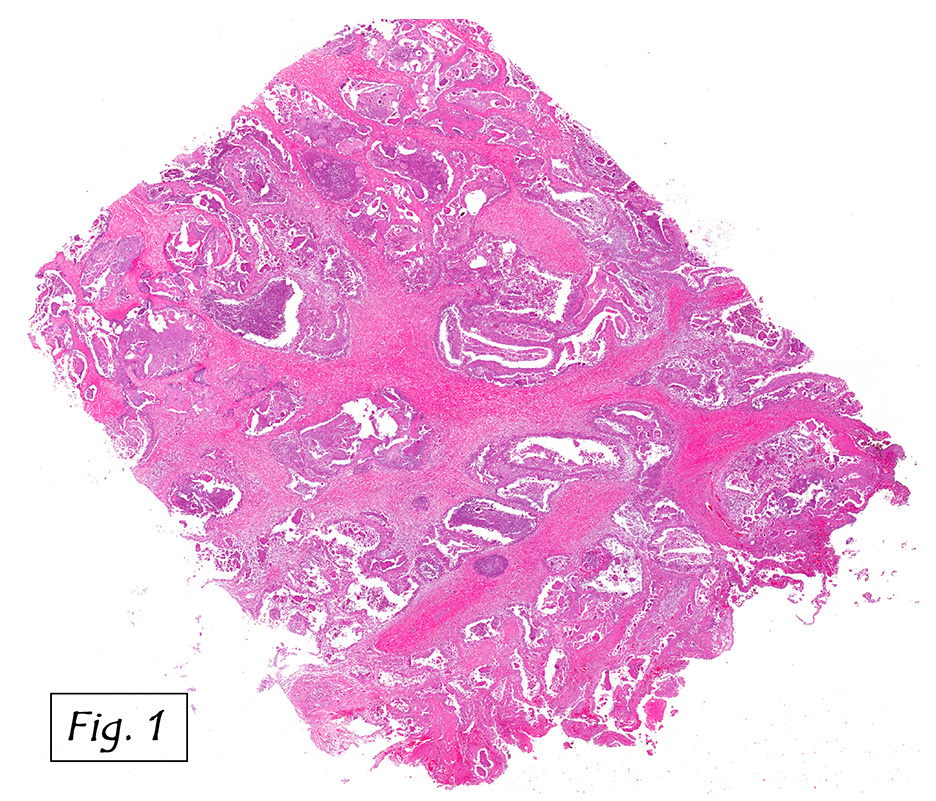
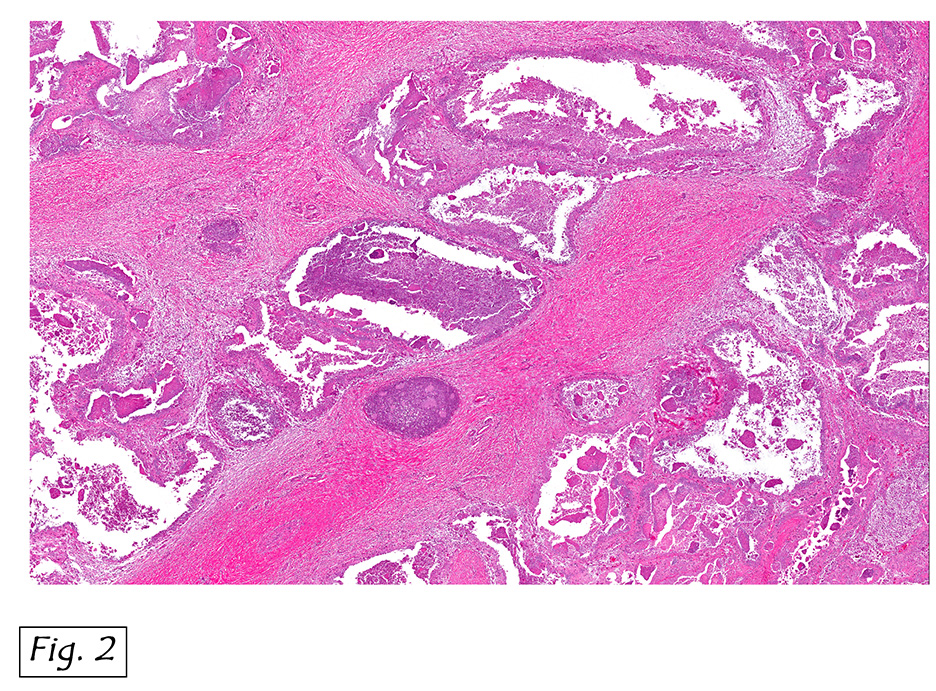
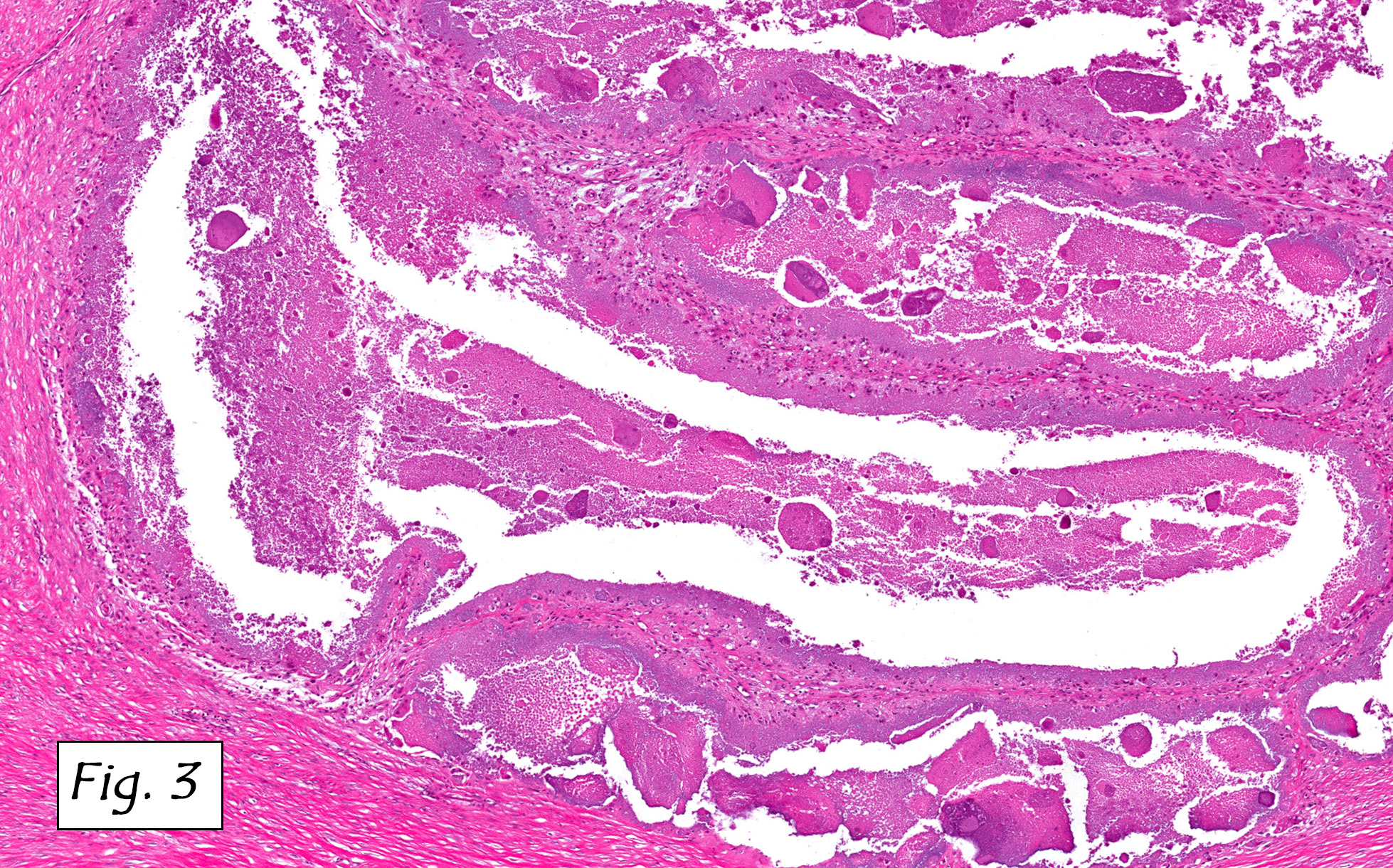
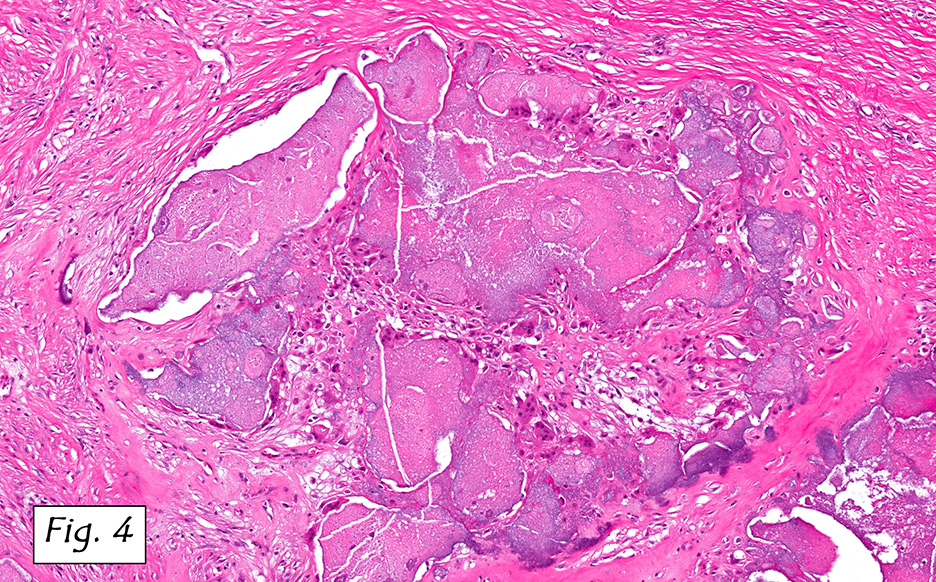
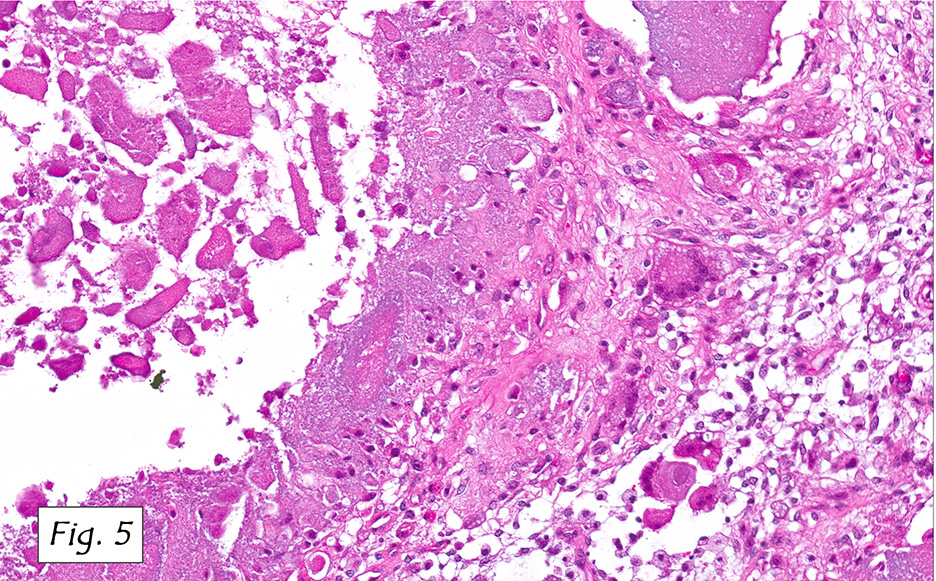
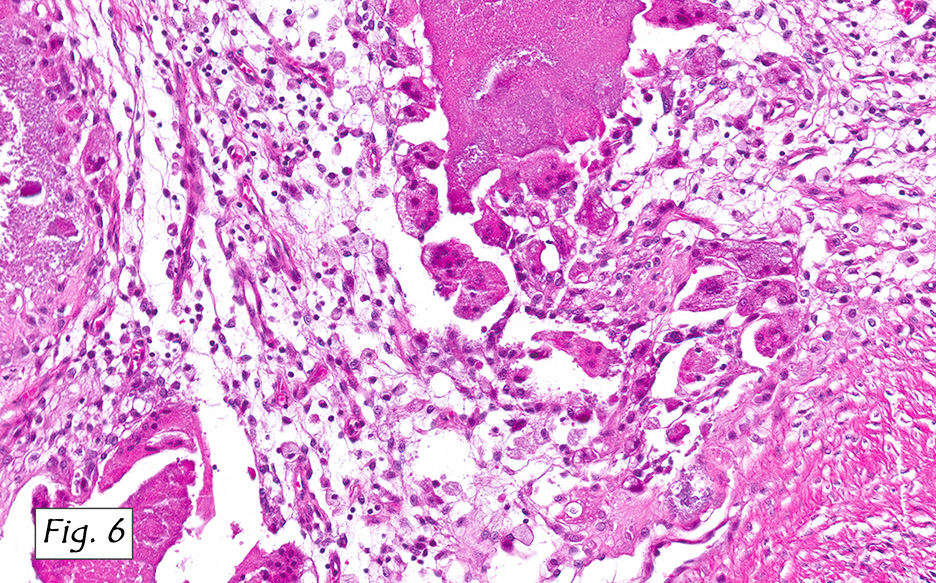
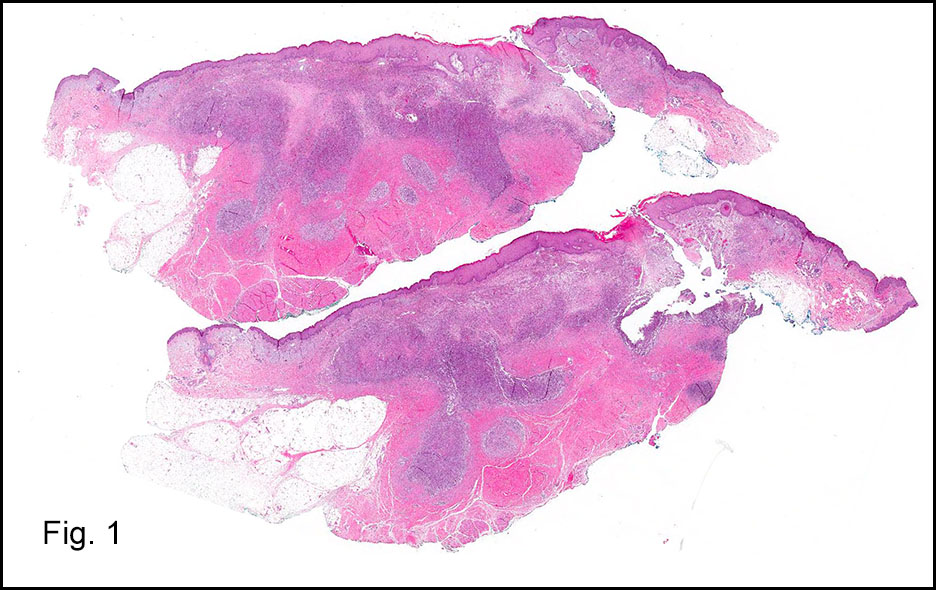
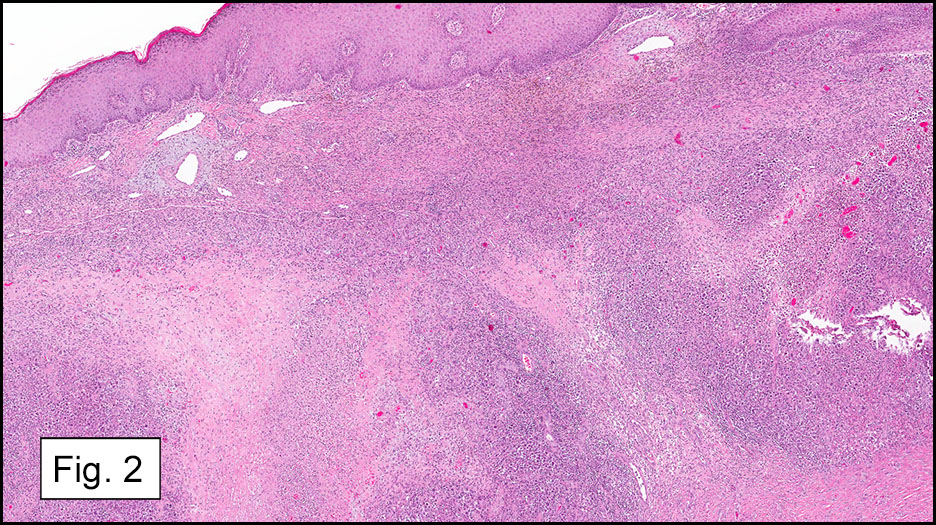
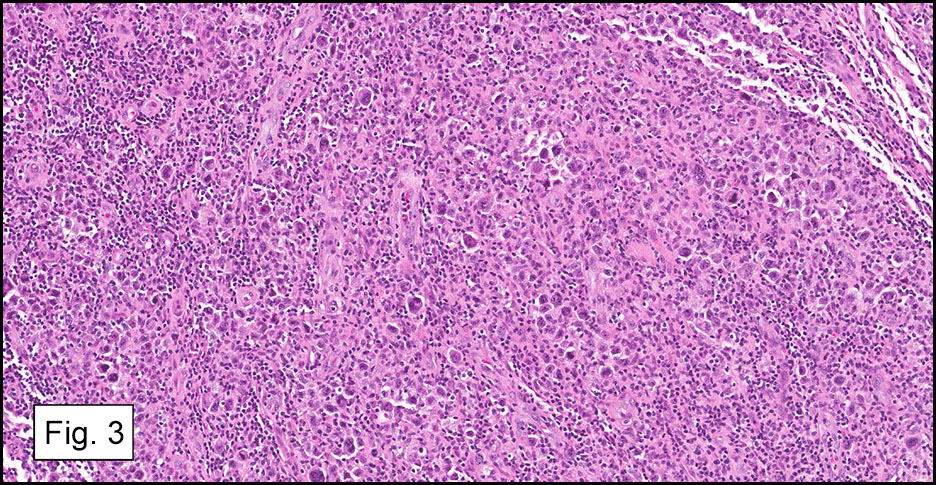
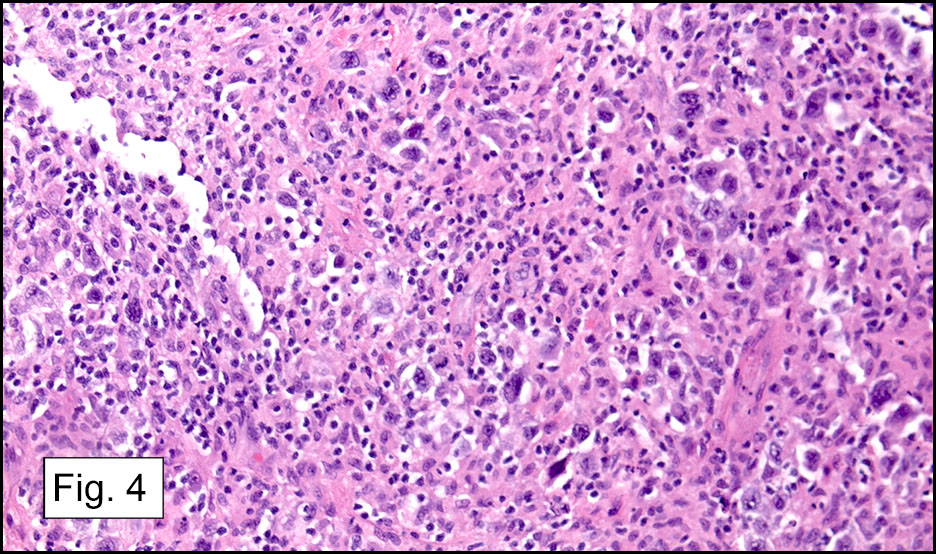
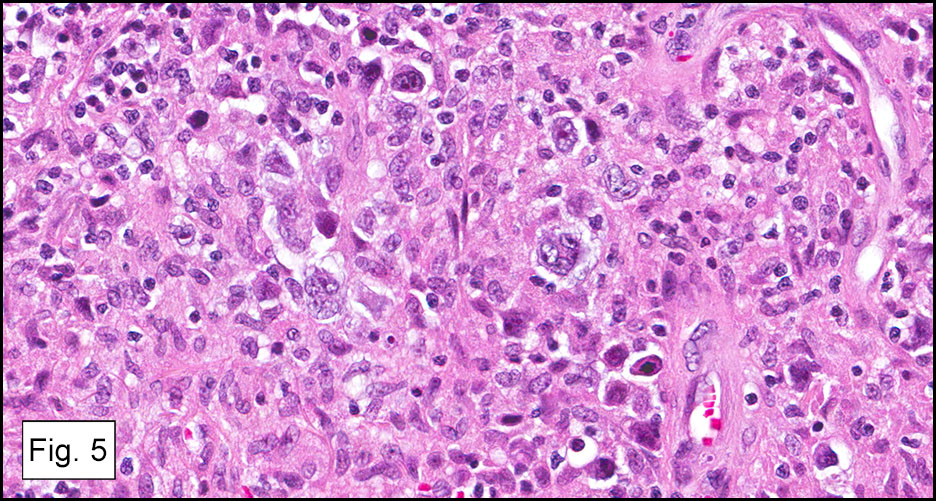
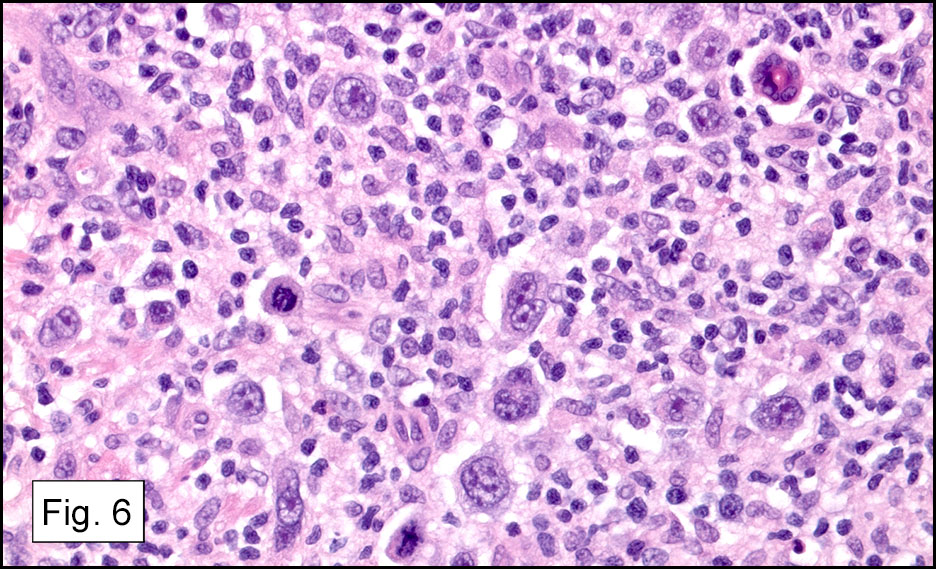
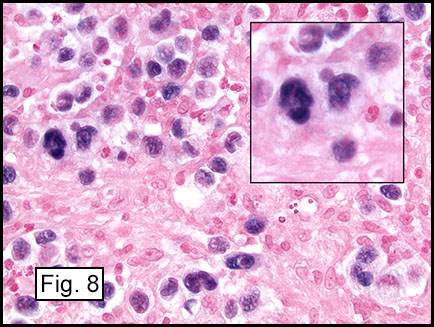
Microscopically, the adrenal glands were necrotic and focally hemorrhagic (Fig. 1). Higher magnification revealed multiple organisms, including intracellular organisms within macrophages (Figs. 2, 3), some of which were adjacent to the areas of necrosis (Fig. 4).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Diagnosis: Adrenal Histoplasmosis
Dennis Aaron Reinke, M.D. and Donald R. Chase, M. D.
Department of Pathology and Human Anatomy,
Loma Linda University Medical Center, Loma Linda, California
California Tumor Tissue Registry, Loma Linda, California
Discussion: Histoplasmosis is caused by the fungus Histoplasma capsulatum. It can be seen in many countries including the United States where it usually occurs in the eastern and central regions. It is transmitted by spore inhalation which can result in a pulmonary infection. It can disseminate to other organs via the blood to affect the spleen, liver, lymph nodes, bone marrow and adrenal glands.
There are a number of causes of adrenal insufficiency which can be categorized as either primary or secondary. Primary causes to consider include infectious, autoimmune, bilateral adrenal hemorrhage (resulting from sepsis, etc.), drug-induced, certain genetic disorders, adrenal infiltration (resulting from metastases, amyloidosis, etc.), or bilateral adrenalectomy. Secondary causes would include isolated adrenocorticotropic hormone deficiency, prior chronic glucocorticoid excess (which may be due to Cushing’s syndrome or steroid therapy), hypothalamic-pituitary region tumors, or pituitary apoplexy.
The clinical presentation of adrenal histoplasmosis may include weight loss, chronic fatigue, fever and anorexia. Primary adrenal insufficiency/Addison’s disease may also manifest in 5-71% of cases, and it is the most frequent cause of fatality. In these cases symptoms may include malaise, fever, nausea, vomiting and orthostatic hypotension. Lab values may show eosinophilia, hyponatremia and hyperkalemia. Imaging studies can reveal symmetrical adrenal enlargement with areas of hemorrhage and necrosis. These findings would suggest a differential diagnosis of aspergillosis, tuberculosis, or other infections, but a definitive diagnosis of adrenal histoplasmosis is not made by imaging studies.
In immunocompromised individuals, the fungus develops in phagocytic cells and macrophages. In patients that are immunocompetent, granulomas may develop that appear similar to noncaseous tuberculosis.
Microscopically, intracellular Histoplasma can be seen on H&E-staining. However, in necrotic and epithelioid cell granulomatous lesions, special stains (i.e. GMS) may be necessary.
Yeast in these tissue sections are around 3 µm in diameter, may be grouped, and have a single bud connected by a narrow base. These organisms have a firm cell wall, and during fixation the protoplasm withdraws from this firm wall. This leaves a resulting clear space that presents the false effect of an unstained capsule in these organisms.
The differential may include Leishmania, Toxoplasma, Blastomyces and Cryptococcus. Leishmania may look very similar on low power, but higher magnification can show the kinetoplast of these organisms. Toxoplasma is generally smaller and not identified within macrophages. Blastomyces have two or more nuclei, while Histoplasma has a single nucleus. Cryptococcus can be differentiated from Histoplasma, as the former’s walls should stain with carmine dyes.
Suggested Reading:
Arlt W, Allolio B. Adrenal insufficiency. The Lancet. 2003;361(9372):1881-1893.
Binford, CH, Dooley, J. Histoplasmosis. In: Binford, CH, ed., Conner, DH, ed. Pathology of Tropical and Extraordinary Diseases. Volume 2. Washington, D.C.: Armed Forces Institute of Pathology; 1976:578-580.
Larbcharoensub N, Boonsakan P, Aroonroch R, et al. Adrenal histoplasmosis: a case series and review of the literature. Southeast Asian J Trop Med Public Health. 2011;42(4):920-5.
Vyas S, Kalra N, Das PJ, et al. Adrenal histoplasmosis: An unusual cause of adrenomegaly. Indian J Nephrol. 2011;21(4):283-5.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}