History: A 28-year-old male presented with a left epigastric mass that had been present for ten years. During exploratory surgery, a 15 cm mass was excised from the greater omentum. It was gray-tan with a heterogeneous surface.
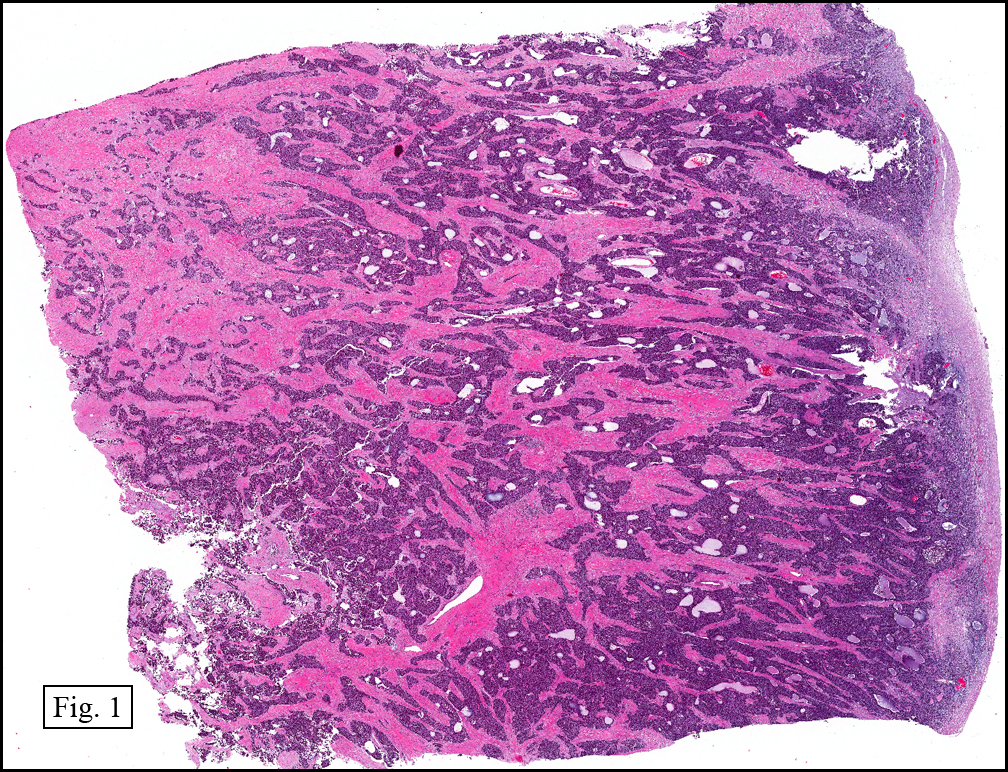
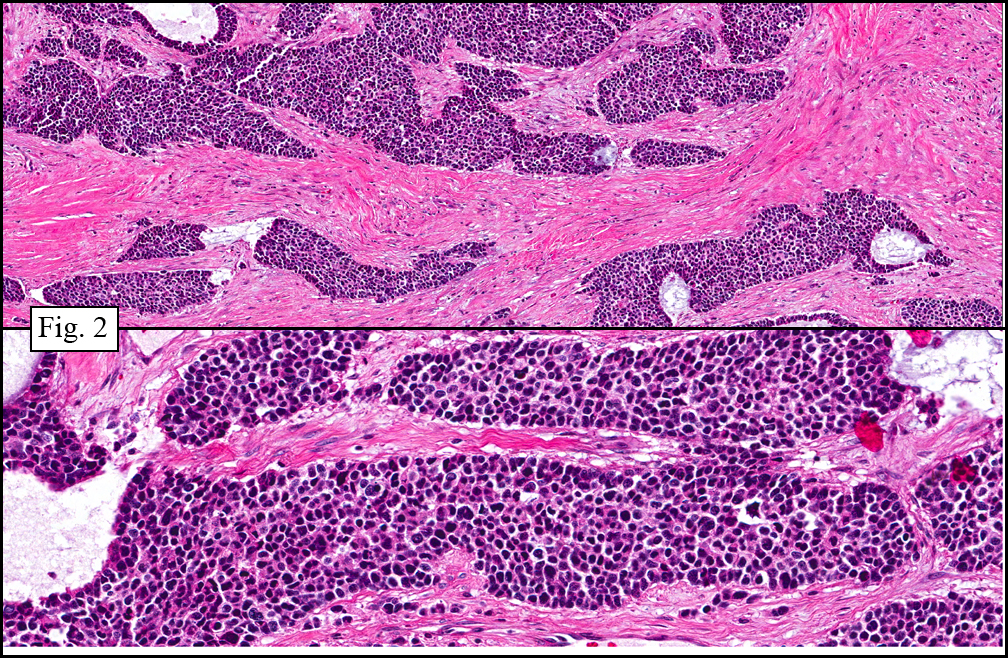
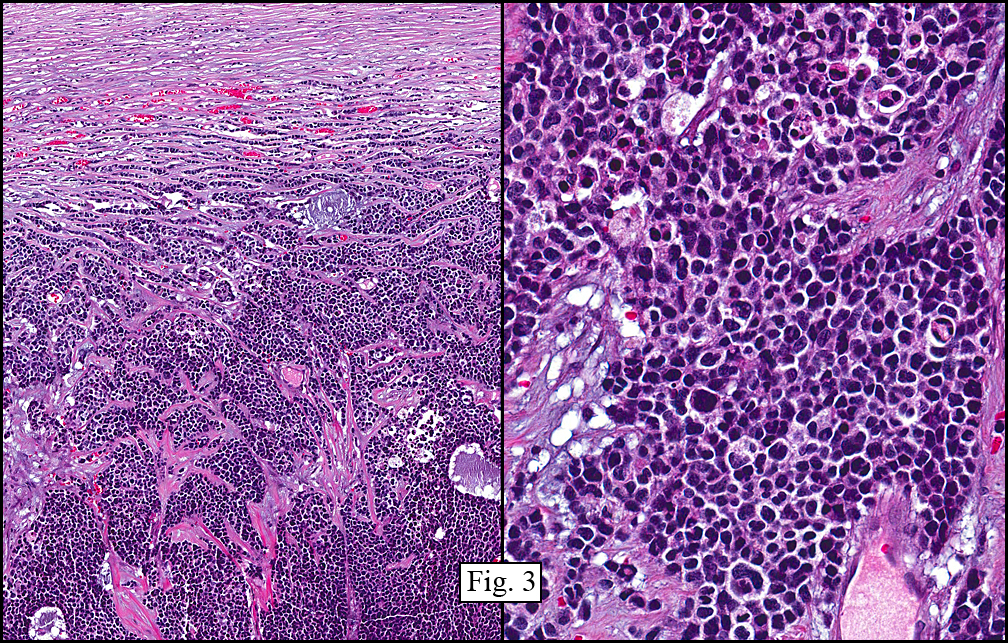
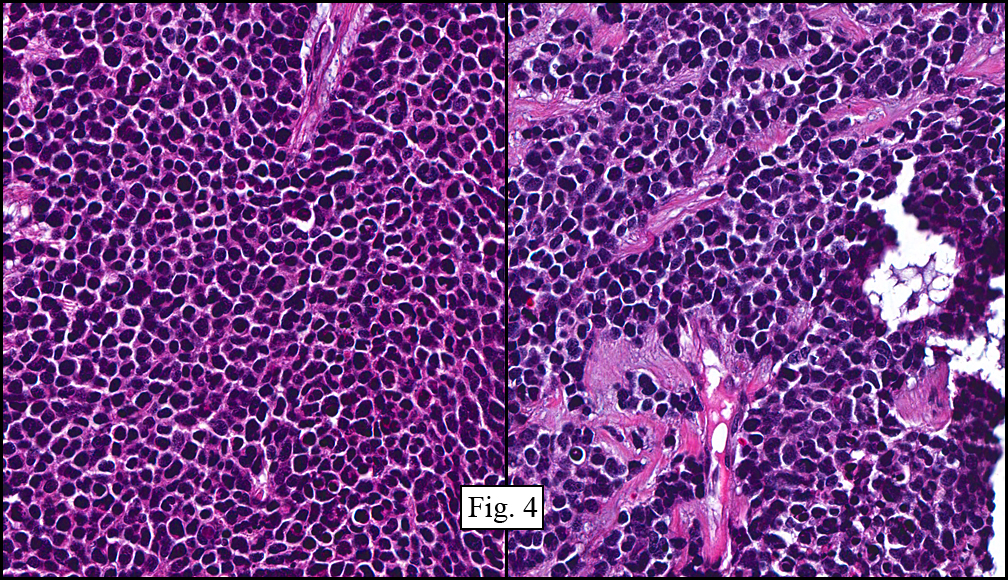
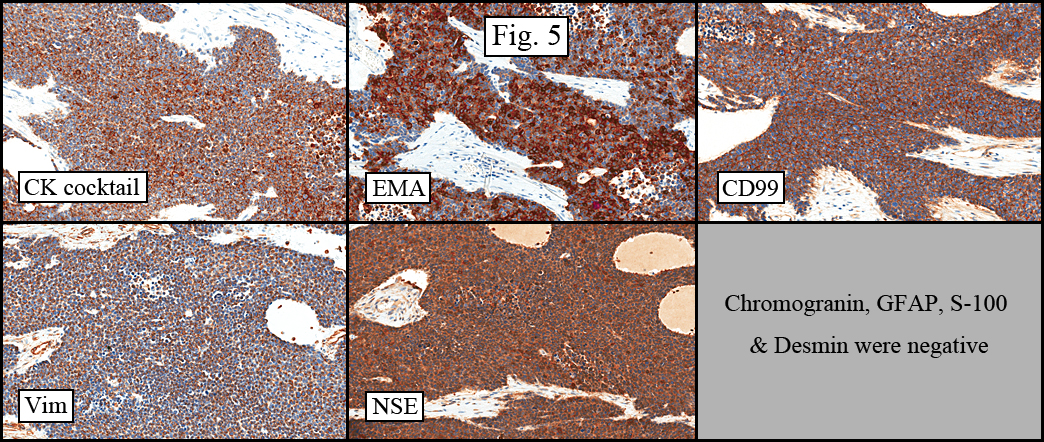
The tumor showed a peculiar trabecular growth pattern (Fig. 1), consisting of cohesive, small, polygonal to round blue cells, sometimes nested, with a prominent supporting desmoplastic stroma (Fig. 2). At the periphery of the tumor the tumor grew in nests and single files (Fig. 3). A subtle but substantial vascular network was seen (Fig. 3). There were scattered mitotic figures and mild pleomorphism (Fig. 3, 4). The tumor displayed a polyphenotypic immunoprofile and had positive staining for cytokeratin, EMA, CD99, Vimentin and NSE. It was negative for chromogranin, GFAP, S-100 and desmin (Fig. 5).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Diagnosis: “Desmoplastic Small Round Cell Tumor of Abdomenâ€
Miriam Peckham, PSF, and Donald R. Chase, MD
Department of Pathology and Human Anatomy, Loma Linda University and
Medical Center, Loma Linda, California
California Tumor Tissue Registry, Loma Linda, California
Discussion: First described about 20 years ago by Gerald and Rosai, desmoplastic small round cell tumor (DSRCT) is a rare malignant lesion presenting in the abdomen with distension and discomfort. It is usually peritoneal-based. DSRCT is typically found in young males (4:1) and occurs between the ages of 15 and 35. Though this tumor presents most commonly in the abdomen and pelvis, it has also been documented in various non-peritoneal sites including the parotid gland, CNS, pancreas, ovary, and lung.
Because of its lack of a definite visceral origin (though there is substantial evidence that it arises from mesothelial cells), this tumor is virtually undifferentiated and has a microscopic appearance of primitive, small, round, nested blue cells (Zellballen-like) within a desmoplastic stroma. Though this microscopic pattern is noted in a large portion of tumors, the histology may vary significantly. Some DSRCTs may show unusual cytology with spindle, rhabdoid, signet-ring, and pleomorphic cells.
Another diagnostic characteristic of DSRCT is its tendency to immuno-mark for neural, mesenchymal, and epithelial elements. The great majority of DSRCT cases stain positively for epithelial markers such as EMA and cytokeratins, as well as mesenchymal markers such as vimentin and desmin (usually in a dot-like, perinuclear pattern). Desmin positivity, which is frequently considered a diagnostic marker, was not noted in our presentation case. DSRCT has also been shown to have multiple neural antigens with neuron-specific enolase being the most common. Chromogranin and synaptophysin positivity, however, is not common. A more specific characteristic of this lesion is that it expresses WT1 – the Wilms’ tumor gene. This is believed to be a central component of DSRCT’s pathogenesis.
An important step in diagnosing DSRCT is separating it from similar-appearing small round blue cell tumors including rhabdomyosarcoma, PNET, Wilms tumor, lymphoma, neuroblastoma, small cell carcinoma, poorly differentiated carcinoma, malignant mesothelioma, and Merkel cell carcinoma. These tumors can be distinguished from DSRCT based on location (whether intra or extra-abdominal), age group (pediatric to young adult patients), and most convincingly, staining patterns via immunohistochemistry. Because of DSRCT’s characteristic polyphenotypia, an immunostain panel is needed to distinguish this entity from similar appearing tumors.
The most defining diagnostic feature of DSRCT is a reciprocal translocation of chromosomes 11 and 22, more specifically t(11;22)(p13;q12). This translocation exhibits the same genes found in Wilms tumor and Ewings sarcoma – fusion of the WT1 gene, from chromosome 11, and the EWS gene from chromosome 22. Other breakpoints for this translocation have been documented as well. Involvement of the WT1 protein has fueled speculation that its overexpression is related to the expression of both mesenchymal and epithelial markers. The EWS-WT1 chimeric protein has also been found to activate, through transcription factors, insulin-like growth factor 1. This is a possible mechanism for proliferation of tumor cells.
DSRCT behaves very aggressively and has a less than 2 year average survival rate. In the abdominal cavity it progressively fills the region with neoplastic nodules and accompanying ascites. These multiple implants frequently make complete excision impossible. A study done in 1998 followed up on 35 patients with DSRCT and found that 25 of those patients had already died, succumbing to widespread metastasis. The remaining patients lived on for an average of 25.2 months. Though the outlook of this disease is grim, the combination of surgical debulking and multimodality therapy can possibly improve survival. A study done by Lal & Sue, et al, showed a 55%, 3-year-survival in patients undergoing a combination of surgery, chemotherapy, and radiotherapy.
Suggested Reading:
1. Weiss SW, Goldblum JR. Soft Tissue Tumors. Mosby Inc. through Elsevier Inc. Philadelphia. 2008.
2. Neder L, Scheithauer BW, Turel KE, Arnesen MA, Ketterling RP, Jin L, Moynihan TJ, Giannini C, Meyer FB. Desmoplastic small round cell tumor of the central nervous system: report of two cases and review of the literature. Virchows Arch. 454:431-39, 2009.
3. Hiralal, Gamanagatti S, Thulkar S, Rao SK. Desmoplastic round cell tumour of the abdomen. Singapore Med J. 48(1): e19, 2007.
4. Lee Y, Hsiao C. Desmoplastic small round cell tumor: A clinicopathologic, immunohistochemical and molecular study of four patients. J Formos Med Assoc. 106(10):854-60, 2007.
5. Murphy AJ, Bishop K, et al. A new molecular variant of desmoplastic small round cell tumor: Significance of WT1 immunostaining in this entity. Hum Pathol. 39:1763-70, 2008.
6. Rosai J. Proceedings of the 107th California Tumor Tissue Registry semi-annual seminar, syllabus pages 32-33, December 5, 1999.
7. lal DR, Su WT, Wolden SL, et al. Results of the multimodal treatment for desmoplastic small round cell tumors. J Pediatr Surg. 40:251-5, 2005.
Wed, Aug 11, 2010
Great case. How do you resolve the discrepancy in this case with the usual natural progression of these tumors? If this followed the usual course for DSRCT, wouldn’t this patient have died long before its resection, ten years after its discovery?
Thanks!
~Chris
Fri, Aug 20, 2010
I agree with your observation. Some tumors just “don’t read the book”. This tumor may have had a “parent” tumor that degenerated into this polyphenotypic neoplasm, which only then would have the aggressive biologic behavior that we expect.
Sat, Mar 1, 2014
somethings are truly unusual
a) very long history
b) single large mass rather than multiple nodules
c) mucoid areas in the tumor
d) lack of desmin expression.
e) WT 1 staining was not done. (Remember it requires WT1 clones with carboxyl group).
Sadly, I don’t know the D/D.