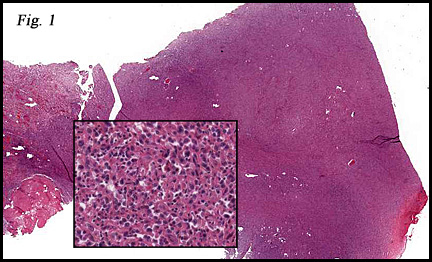
 History: A 28 year-old woman presented with a palpable mass in the deep tissues of the left thigh. A 4.6 x 3.5 x 2.8 cm ovoid rubbery soft tissue mass was removed. Its perimeter was well-circumbscribed and was partially covered by gray-brown fragments of skeletal muscle. The cut surface was glistening and pink-white. Rare microcysts were seen.
History: A 28 year-old woman presented with a palpable mass in the deep tissues of the left thigh. A 4.6 x 3.5 x 2.8 cm ovoid rubbery soft tissue mass was removed. Its perimeter was well-circumbscribed and was partially covered by gray-brown fragments of skeletal muscle. The cut surface was glistening and pink-white. Rare microcysts were seen.
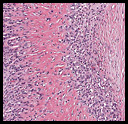
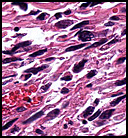
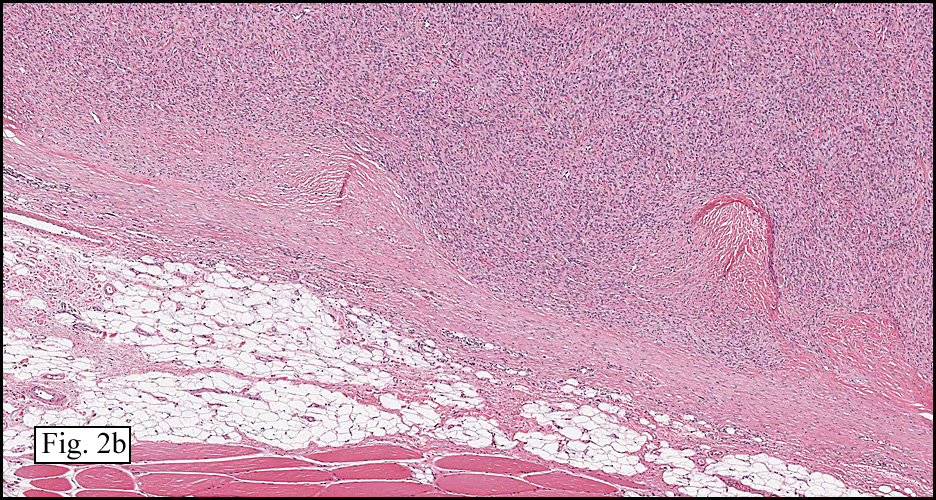
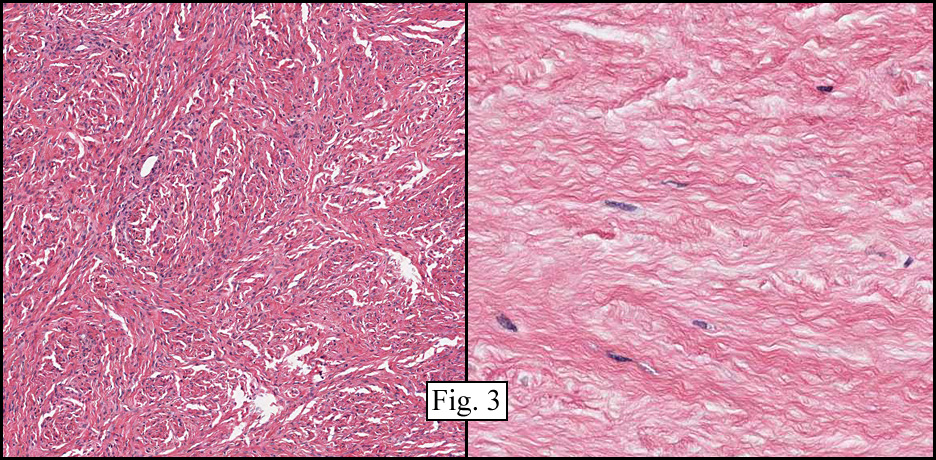
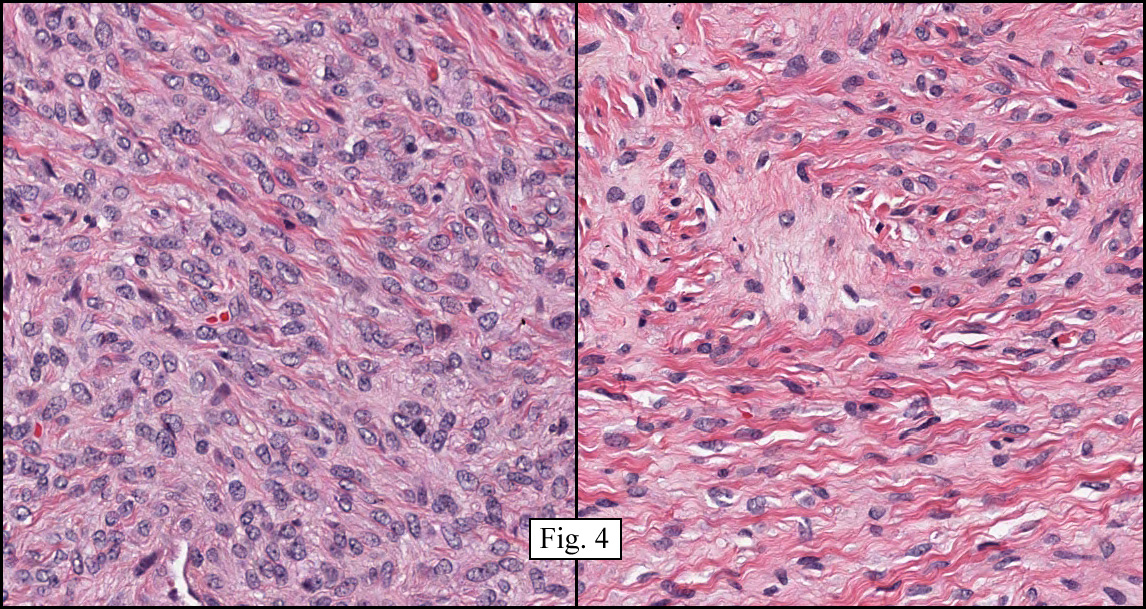
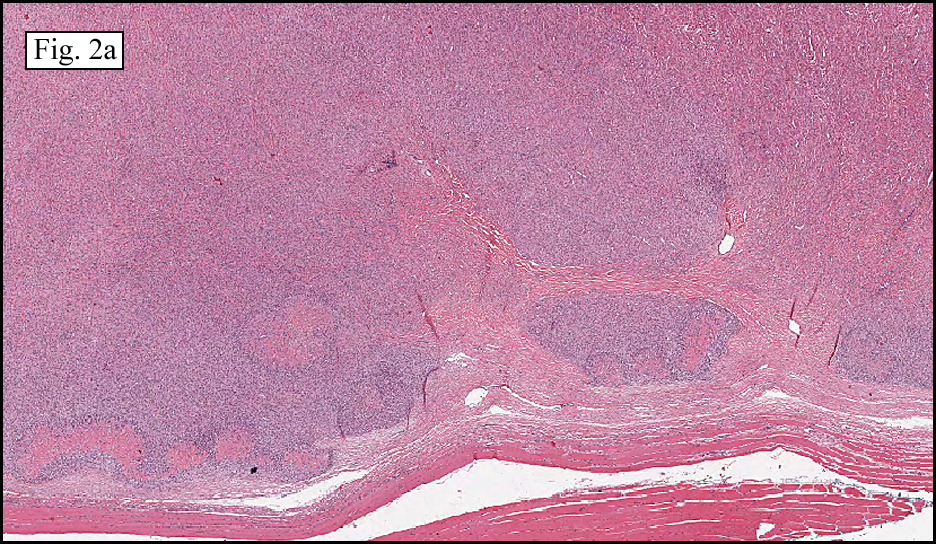
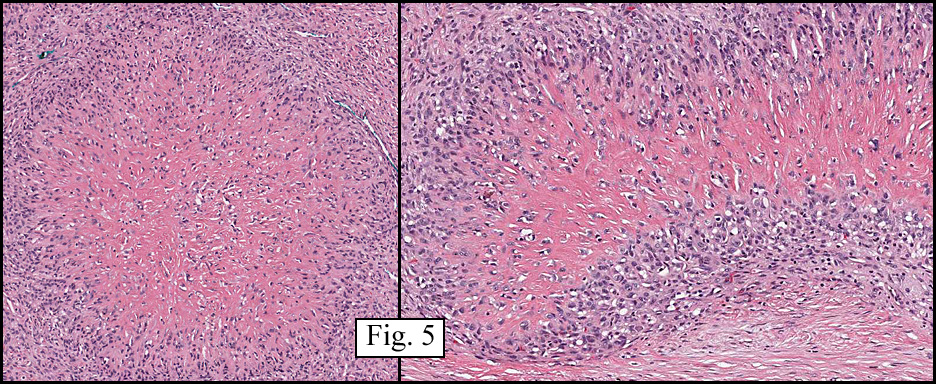
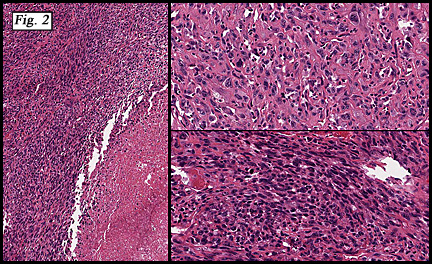
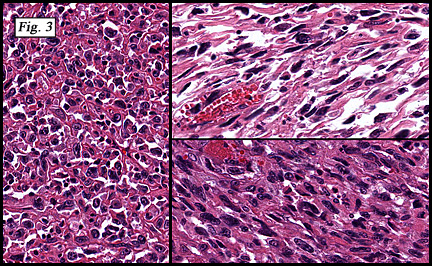
The tumor was sharply demarcated from surrounding skeletal muscle and had a variably thick capsule (Fig. 1–2b). Highly cellular regions were present in close proximity to less cellular, fibrotic regions (Fig. 3). Some regions had a vague storiform growth pattern, but generally the tumor was present in diffuse sheets. Individual cells had round to polygonal nuclei with limited spindling (Fig. 4). Myxoid areas were only infrequently encountered. Low levels of mitotic activity are observed, mostly in the hypercellular regions. Neither atypical mitotic figures nor bizarre nuclear pleomorphism were seen. Of interest were the large, rosette-like structures with hyalinized centers (Figs. 2a, 5).
Diagnosis: “Low-Grade Fibromyxoid Sarcoma With Hyalinizing Giant Rosettesâ€
Timothy R. Smith, MS3 and Donald R. Chase, MD
Department of Pathology and Human Anatomy
Loma Linda University and Medical Center, Loma Linda, California
California Tumor Tissue Registry, Loma Linda, California
Discussion: Low-Grade Fibromyxoid Sarcoma (LGFMS) is a rare, recently-described soft tissue tumor that is now being more completely characterized. First described by Evans in 1987, LGFMS is also known as “Evans tumorâ€. The tumor usually arises in young to middle-aged adults, but has a range from 3 to 78 years. Males are more commonly affected than females. The lower extremities are the most common site, especially the thigh, although the tumor has now been reported in virtually any location. Although Evans original description did not include rosettes, the tumor has subsequently been found to have large, hyalinizing giant rosettes. This variant was subsequently described 10 years later in a paper by Drs. Lane, Shannon and Weiss et.al (1997) as “hyalinizing spindle cell tumor with giant rosettesâ€. Three years later (2000), Drs Folpe and Weiss proposed a unified theory that the two tumors are likely the same, both behaving as low grade sarcomas. Deceptively bland, both Dr. Evans and Dr. Lanes’ tumor types have high recurrence rates and may eventually dedifferentiate into a higher grade sarcoma. In addition to tumor morphologies that are illustrated in this paper (Figs. 1-5), a minority of the cases show increased perivascular cellularity, moderate nuclear pleomorphism (more often in recurrent tumors), and, in myxoid areas, a rich capillary vascular network.
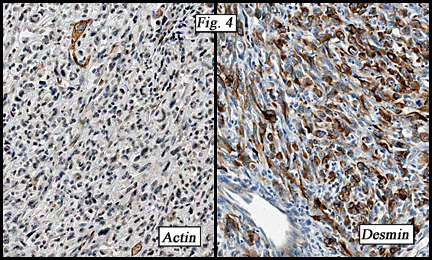
LGFMS is routinely positive for vimentin and may be focally positive for smooth muscle actin. Decoration for S-100 is uncommon but may occur in regions adjacent to rosettes. The tumor is uniformly negative for CD34, EMA, desmin and cytokeratin.
A translocation occurs in up to 88% of the cases and is usually expressed as t(7;16)(q34;p11) FUS-CREB3L2 fusion gene. Genetics losses of 13q also occur (Kiuru-Kuhlefelt et al., unpublished observations.)
The differential diagnosis of LGFMS includes low grade fibroblastic/myofibroblastic tumors and neoplasms with myxoid characteristics:
• Desmoid-type fibromatosis is also composed of uniform, bland cells with minimal mitotic activity. The major difference, though, is the much more organized fascicular growth of fibromatosis and the usual lack of myxoid regions and alternating zones of cellularity. Large rosettes do not occur in desmoid tumors, and significant pleomorphism is absent. B-catenin is negative in LGFMS but may be positive in fibromatosis.
• Neurofibroma (NF) almost always shows staining for S-100 while LGFMS only shows localized S-100 staining near rosettes. The diffuse form of neurofibroma may have Wagner-Meisner bodies which are lacking in LGFMS, and when NF is associated with van Recklinghausen’s disease, it may assume a plexiform pattern, also non-existent with LGFMS. Finally, the nuclear characteristics of peripheral nerve sheath tumors are uniquely different showing asymmetrical shapes resembling commas, apostrophes, and sine waves.
• Low-grade myxoid MFH (myxofibrosarcoma) has considerably more nuclear atypia and a more extensive myxoid matrix (generally more than 50% of its area). It uniformly lacks rosettes.
• Low-grade bland myxoid lesions can be a problem, since myxoid change may occur in virtually every type of mesenchymal neoplasm. LGFMS is characterized by alternating hypercellular/fibrous and myxoid areas and by the occasional swirling whorled cell growth pattern. Negative immunostains for S100, desmin and cytokeratin may help in excluding myxoid variants of neurofibroma, schwannoma, leiomyoma and mucinous carcinoma.
• Intramuscular and juxta-articular myxomas have much lower cellularity, no organized growth pattern and a paucity of blood vessels/capillaries. Since they have no hypercellular regions, they lack the alternating fibrous and myxoid areas that LGFMS possesses. These myxomas have colorfully been described as being “bags of water.â€
LGFMS is a slow growing tumor with a deceptively benign appearance. Its early recognition as a sarcoma is vital since only complete surgical extirpation is initially adequate to prevent recurrence. Although recurrence and/or metastasis may be expected (Evans’ 1993 study showed 7/12 with distant spread), the tumor is indolent and potentially curable if caught early and completely excised.
Suggested Reading:
Evans HL. Low-grade fibromyxomas. A report of two metastasizing neoplasms having a deceptively benign appearance. Am J Clin Pathol 88:615, 1987
Evans HL. Low-grade fibromyxoid sarcoma. A report of 12 cases: Am J Surg Pathol; 17: 595-600, 1993.
Lane KL, Shannon RI, Weiss SW. Hyalinizing spindle cell tumor with giant rosettes: a distinctive tumor closely resembling low-grade fibromyxoid sarcoma. Am J Surg Pathol 21:1481, 1997.
Folpe AL, Lane KL, Paull G, Weiss SW, et al. Low grade fibromyxoid sarcoma and hyalinizing spindle cell tumor with giant rosettes: a clinicopathologic study of 73 cases supporting their identity and assessing the impact of high grade areas. Am J Surg Pathol 24(10):1353-1360, 2000.
Kempson RL, Fletcher CDM, Evans HL, et al: Tumors of the Soft Tissues. Atlas of tumor pathology. Third Series. 30. 99-104, 2001.
Miettinen M. Malignant and Potentially Malignant Fibroblastic and Myofibroblastic Tumors. Diagnostic Soft Tissue Pathology chap 7:193-194, 2003
Perigny M, Dion N, et al. Low grade fibromyxoid sarcoma: a clinico- pathologic analysis of 7 cases. Ann Pathol 26(6):419-25, 2006.
Matsuyama, A, Hisaoka, M, et al. Molecular Detection of FUS-CREB3L2 Fusion Transcripts in Low-grade Fibromyxoid Sarcoma Using Formalin-fixed, Paraffin-embedded Tissue Specimens. Am J Surg Pathol 30(9):1077-1084, 2006.
Downs-Kelly E, Goldblum JR, Patel RM, Weiss SW, et al: The utility of fluorescence in situ hybridizations (FISH) in the diagnosis of myxoid soft tissue neoplasms. Am J Surg Pathol 32(1) 8-13, 2008.
 History: A 42-year-old woman with a one month history of dry cough was radiographically thought to have a left cardiac myxoma and bilateral hydrothorax. At surgery, however, she was found to have a 7.0 x 6.0 x 4.5 cm gray fleshy solid mass in the left pulmonary vein which extended into the left atrium.
History: A 42-year-old woman with a one month history of dry cough was radiographically thought to have a left cardiac myxoma and bilateral hydrothorax. At surgery, however, she was found to have a 7.0 x 6.0 x 4.5 cm gray fleshy solid mass in the left pulmonary vein which extended into the left atrium. History: A 20-year-old woman presented with chronic jaw pain and swelling in the left cheek. Computerized tomography (CT) scan identified an expansile, multilocular, osteolytic mass in the left lower maxillary sinus (
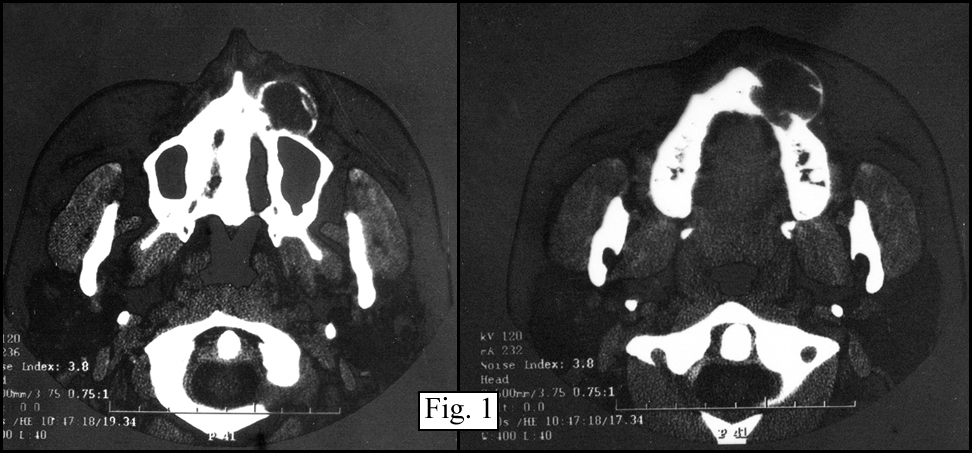
History: A 20-year-old woman presented with chronic jaw pain and swelling in the left cheek. Computerized tomography (CT) scan identified an expansile, multilocular, osteolytic mass in the left lower maxillary sinus ( History: A 28 year-old woman presented with a rapidly growing nodule on the right thumb. Radiographs taken prior to surgery demonstrated an ill-defined soft tissue mass without apparent calcification or periosteal reaction (
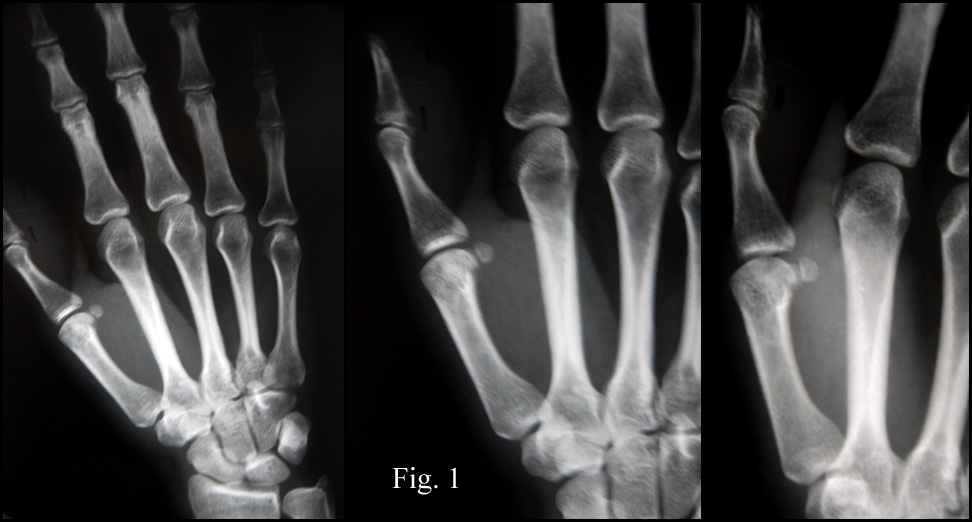
History: A 28 year-old woman presented with a rapidly growing nodule on the right thumb. Radiographs taken prior to surgery demonstrated an ill-defined soft tissue mass without apparent calcification or periosteal reaction ( History: A 71-year-old male presented with persistent headache. CT and MRI scans revealed a 5.0 x 4.0 x 2.5 cm bifrontal brain parenchymal mass with two smaller dural-based satellite lesions measuring 3.0 and 1.0 cm in greatest diameter (
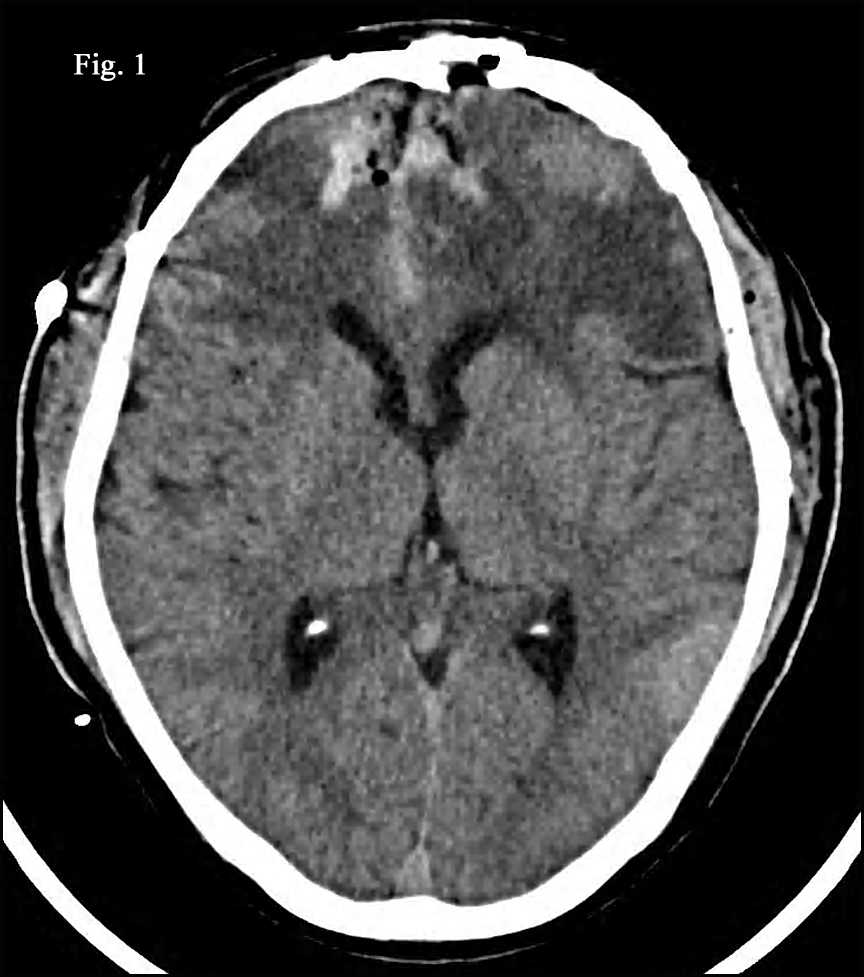
History: A 71-year-old male presented with persistent headache. CT and MRI scans revealed a 5.0 x 4.0 x 2.5 cm bifrontal brain parenchymal mass with two smaller dural-based satellite lesions measuring 3.0 and 1.0 cm in greatest diameter (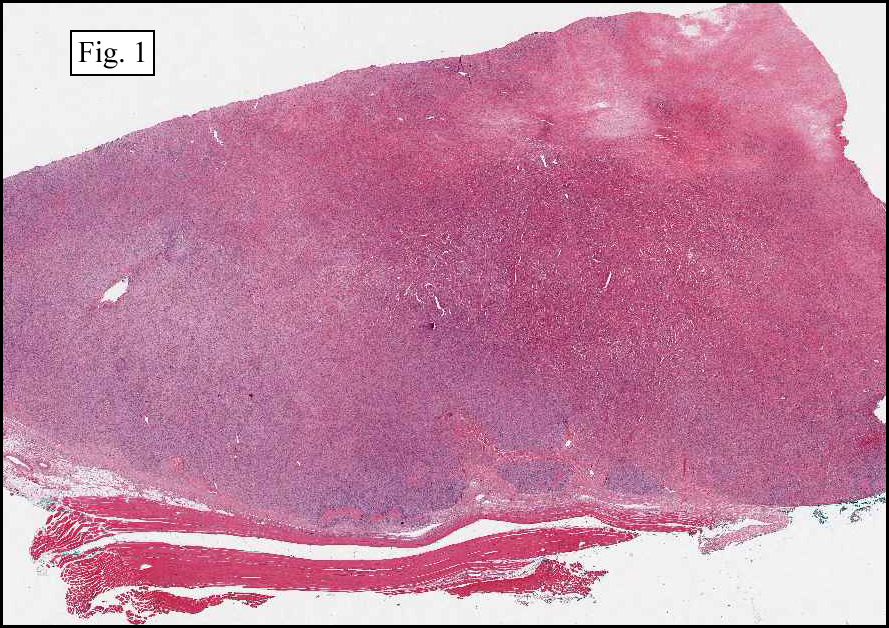{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}