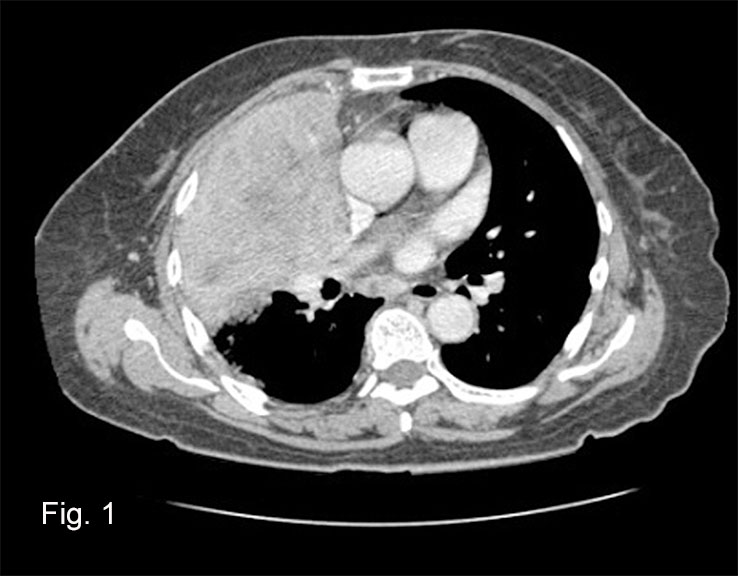
History: A 38 year old woman presented with a several month history of an enlarging painful mass of the right knee. Physical examination showed a firm, tender mass over the medial knee region. An MRI revealed a 6.9 x 4.7 x 3.4 cm, heterogeneously enhancing soft tissue mass with areas of hemorrhage and probable
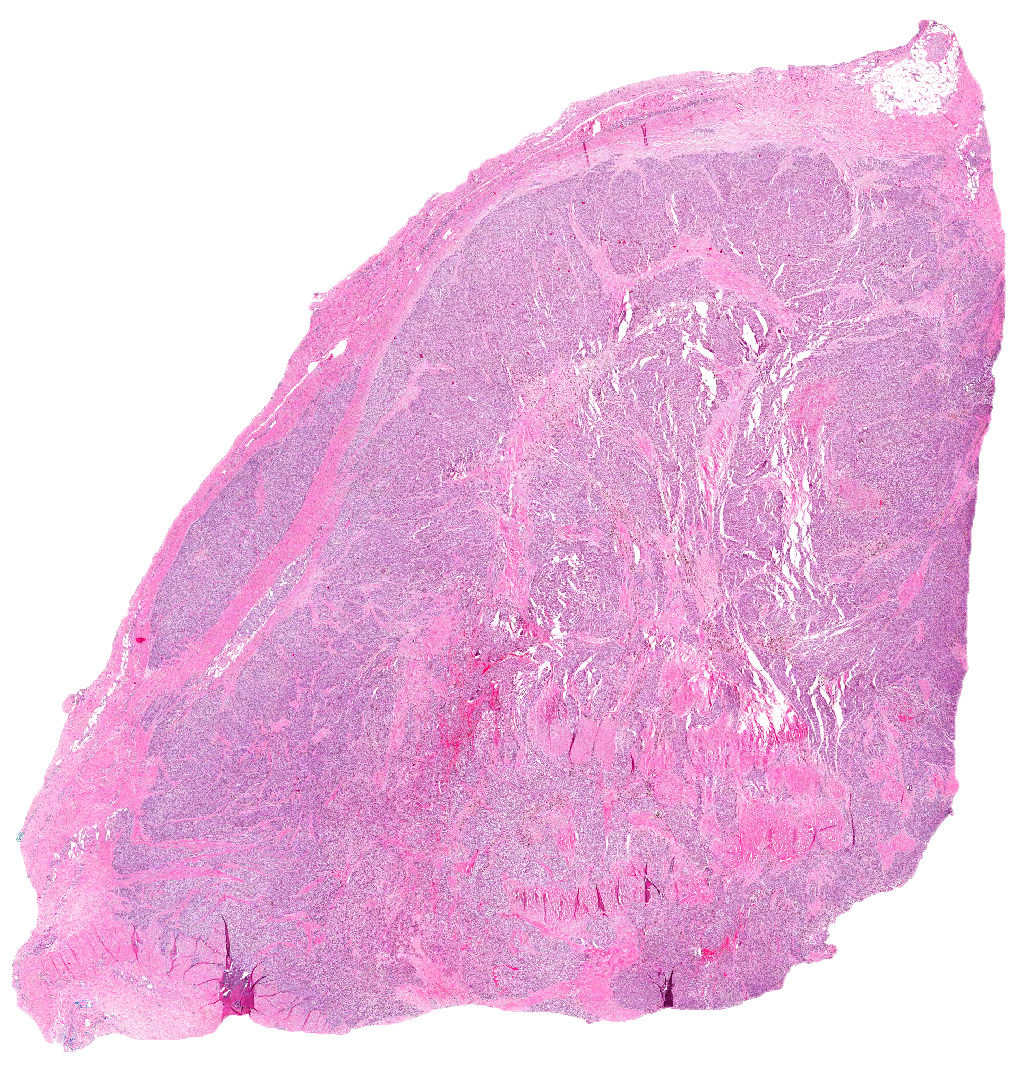
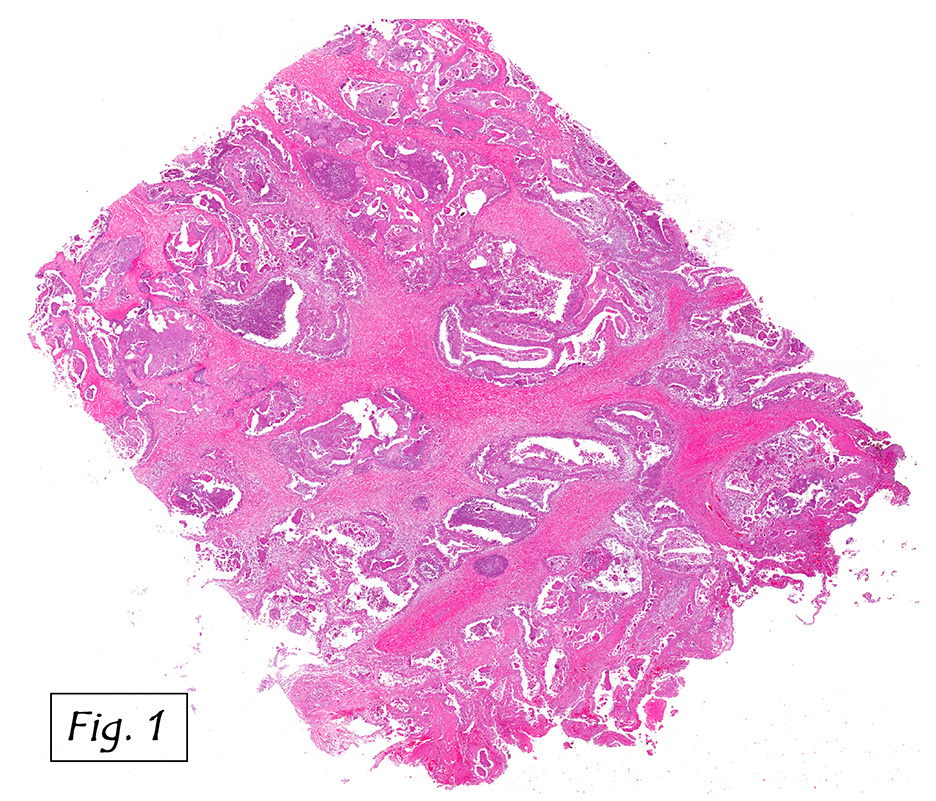
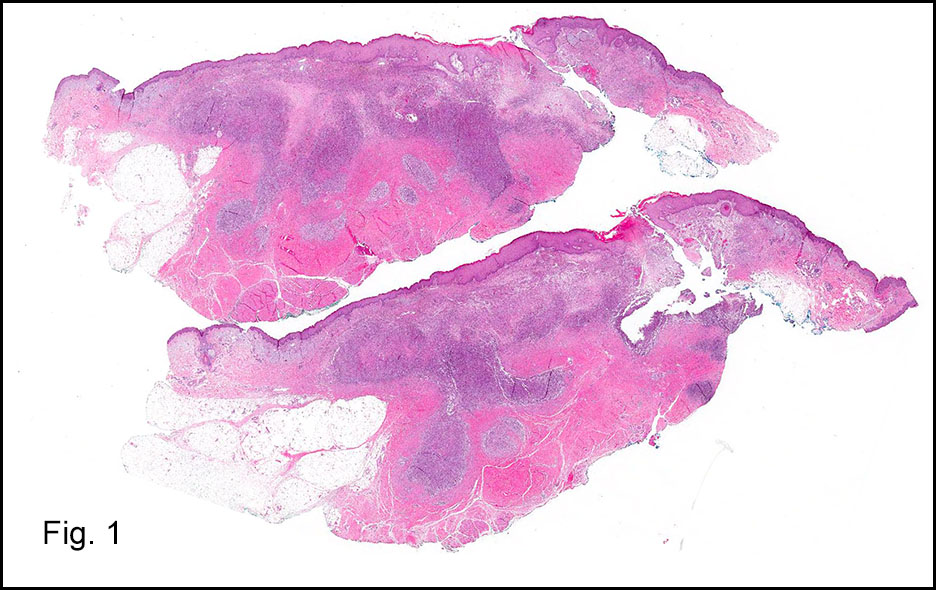
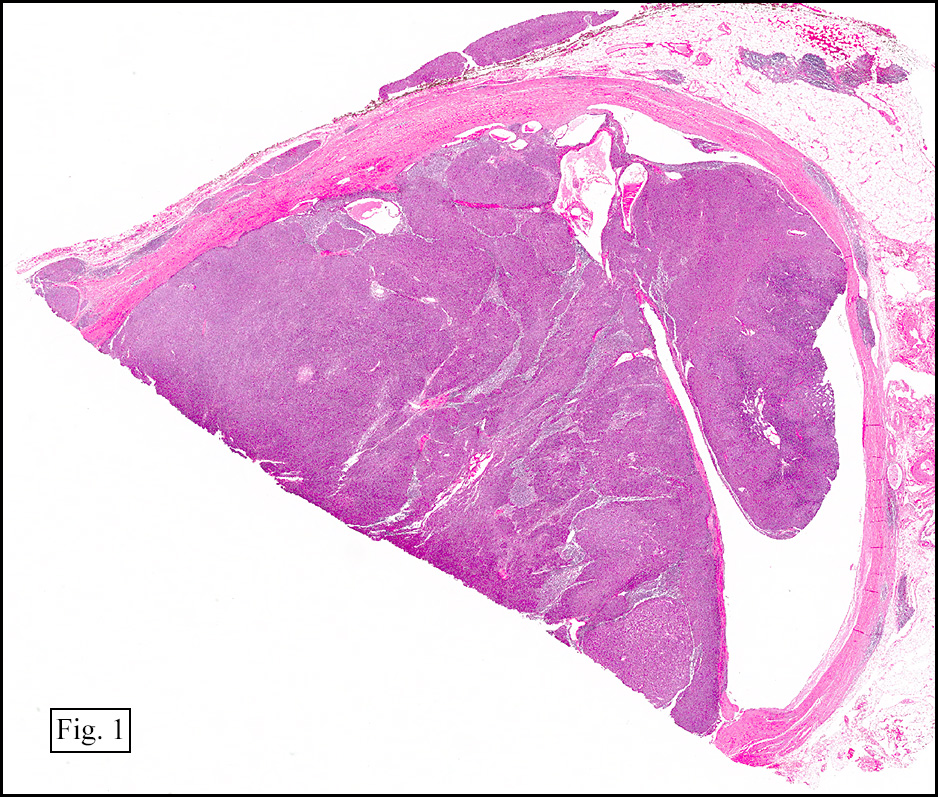
Grossly, the mass was firm, variegated, white-tan to dark-brown, and well-circumscribed (Fig. 1).
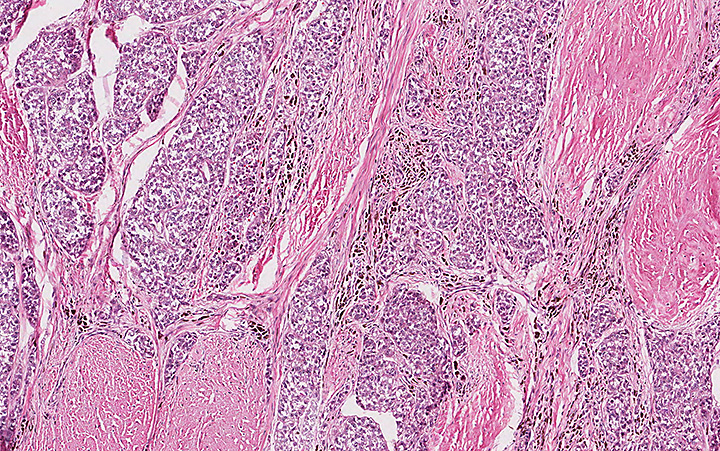
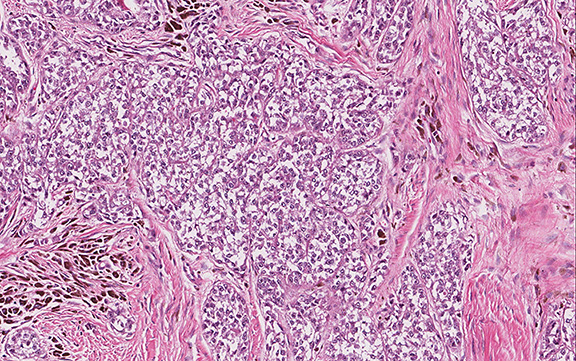
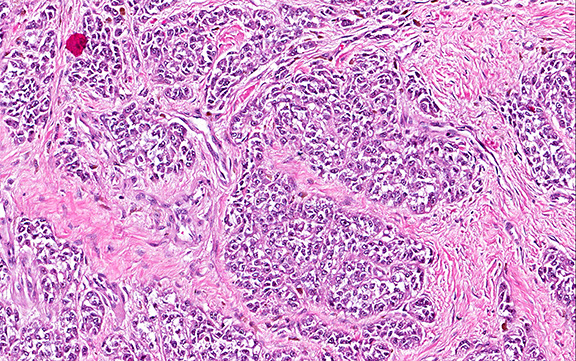
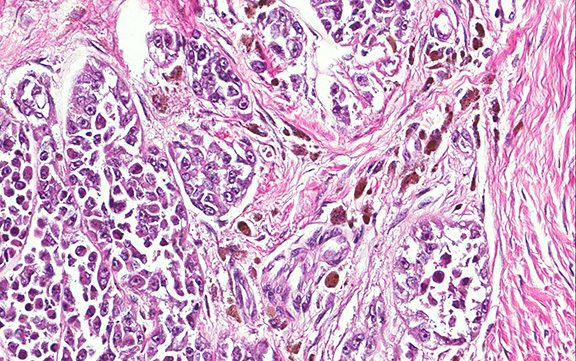
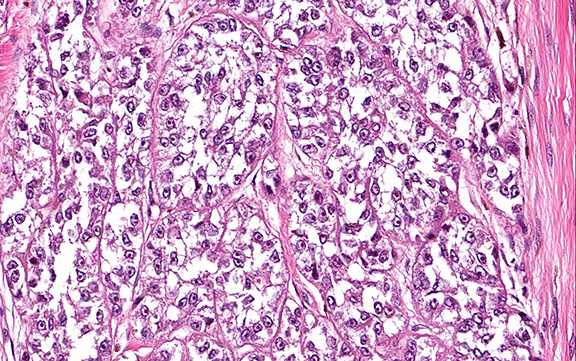
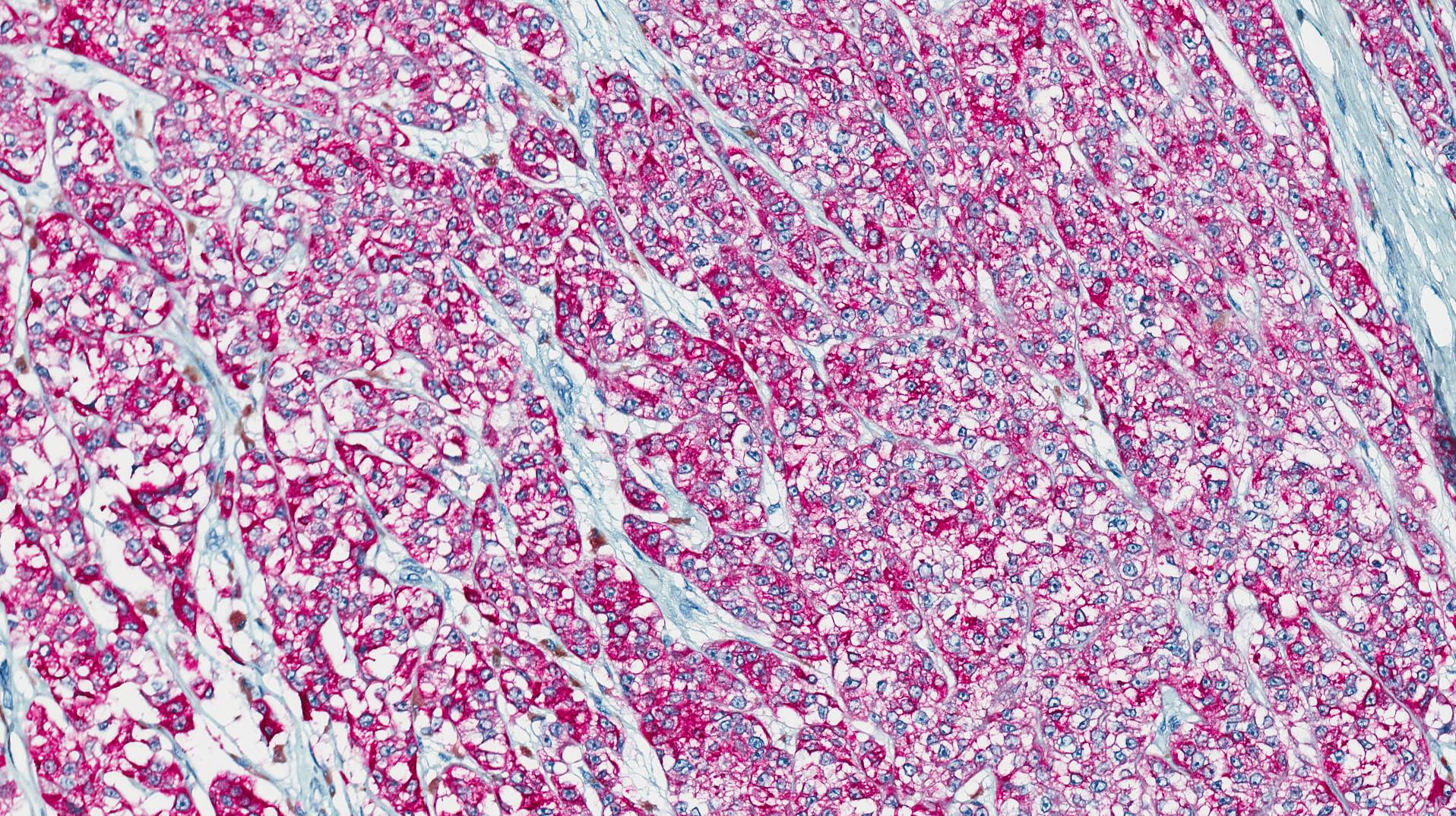
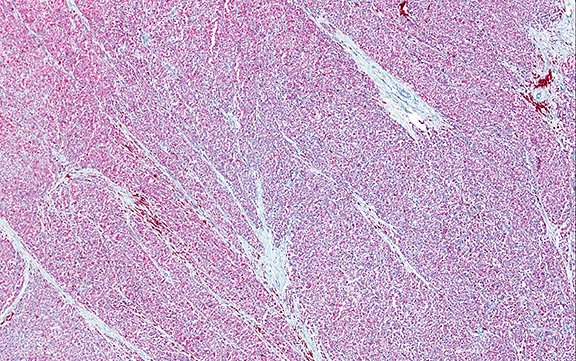
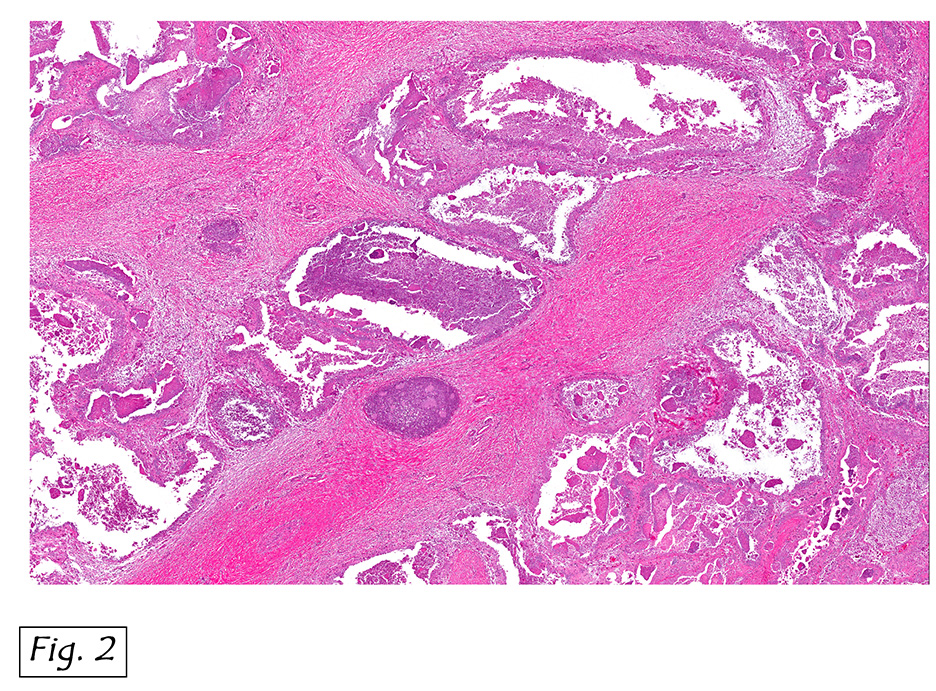
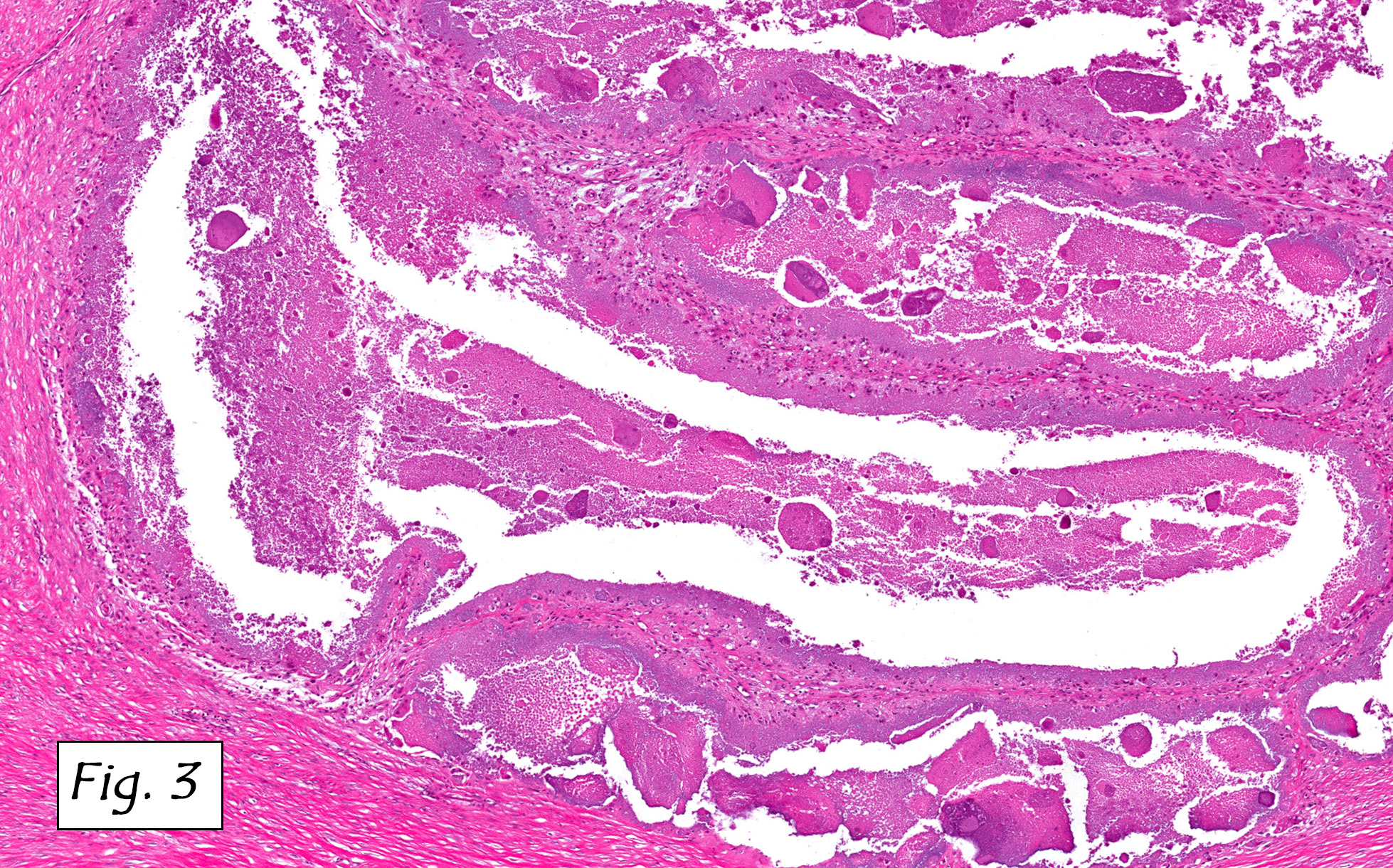
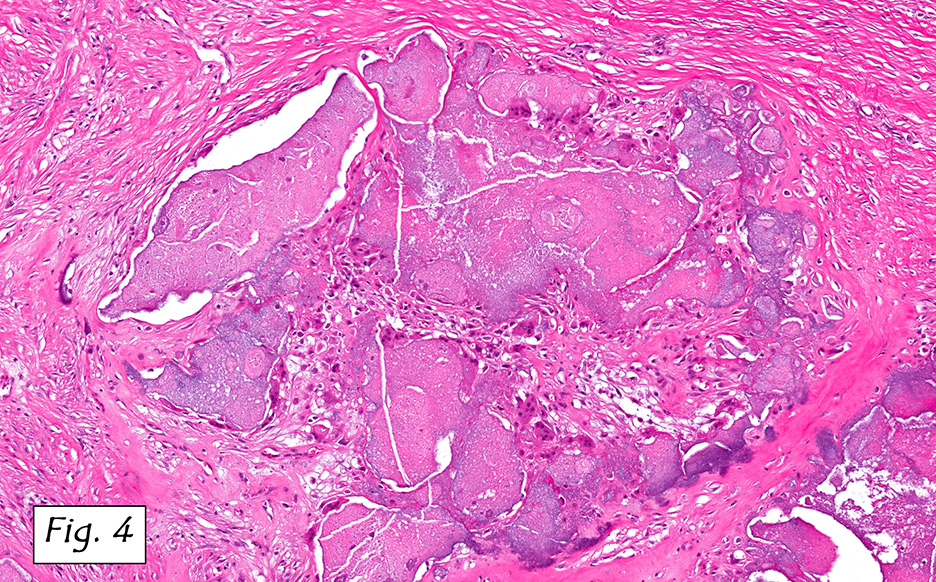
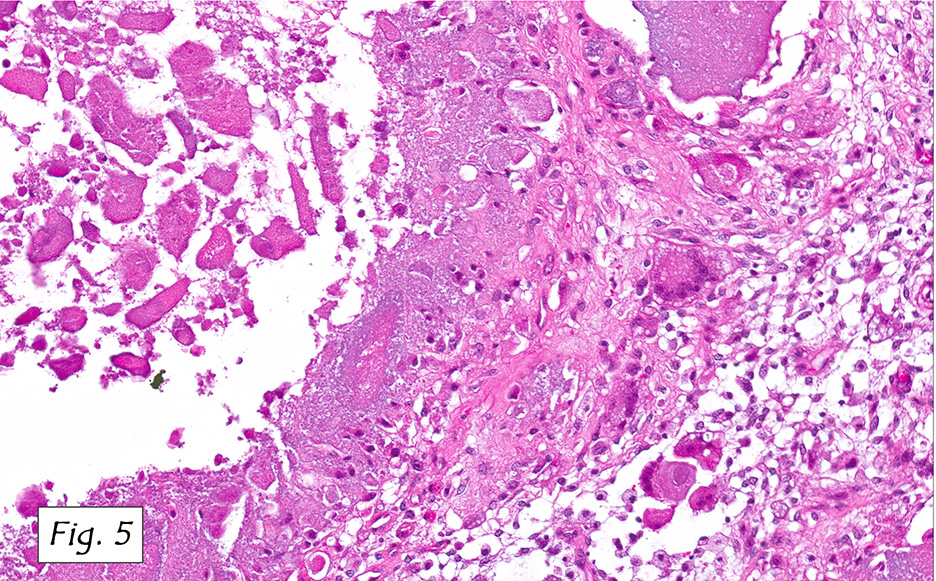
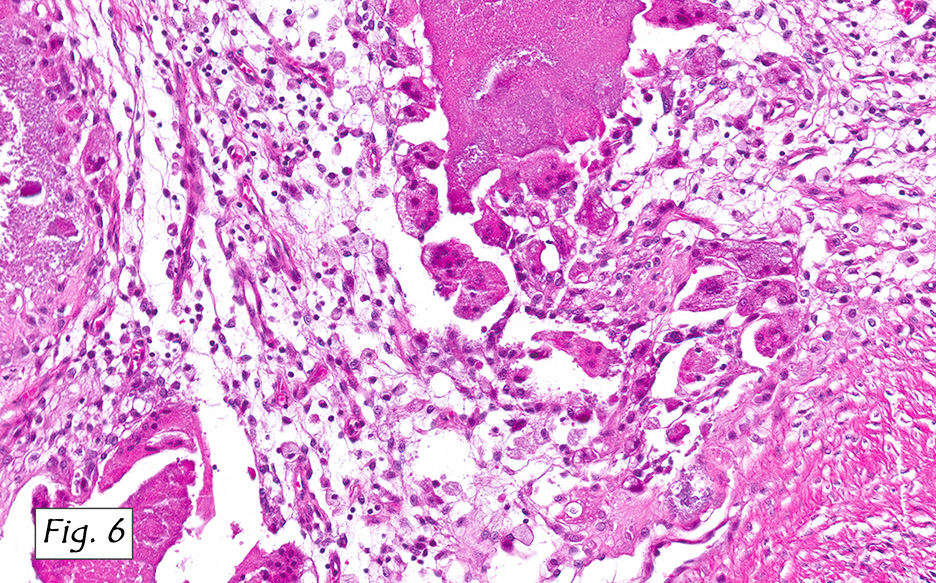
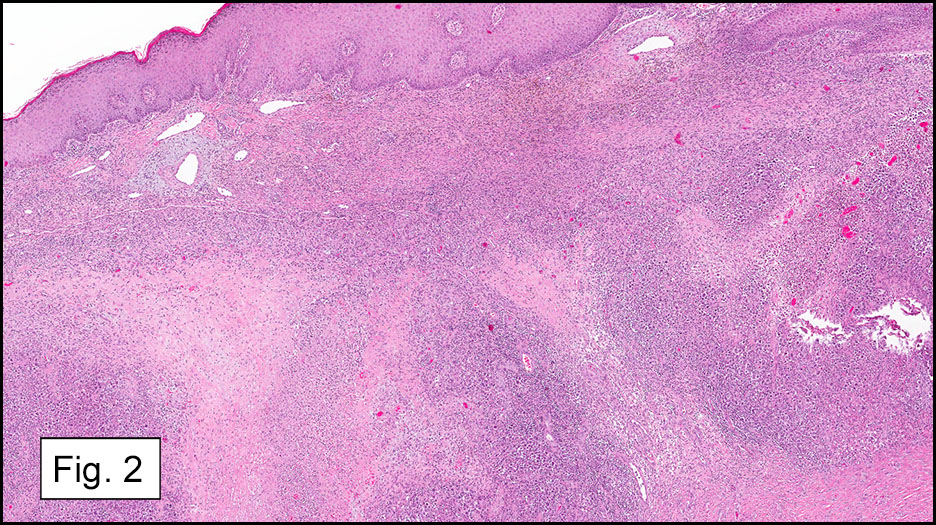
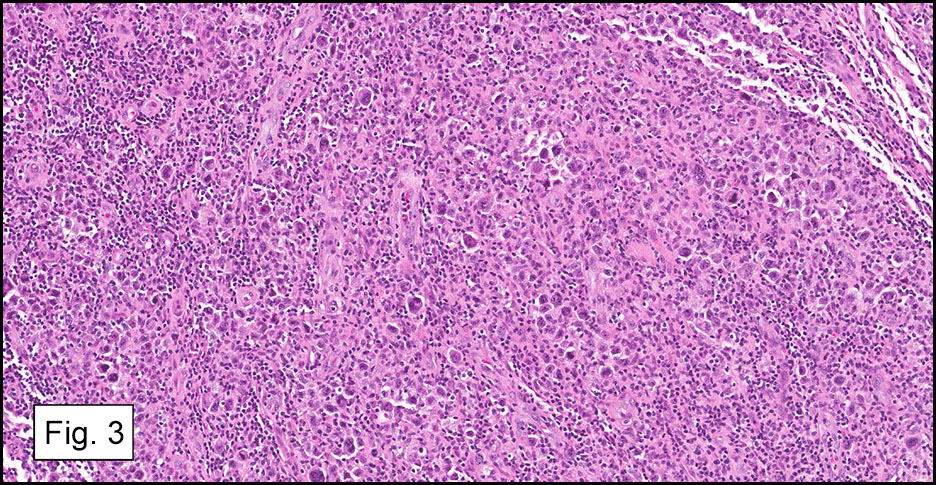
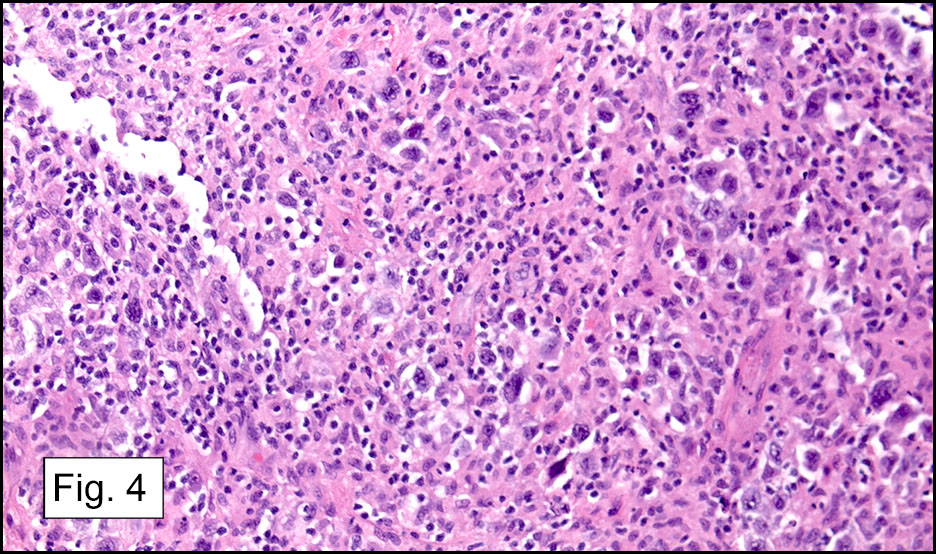
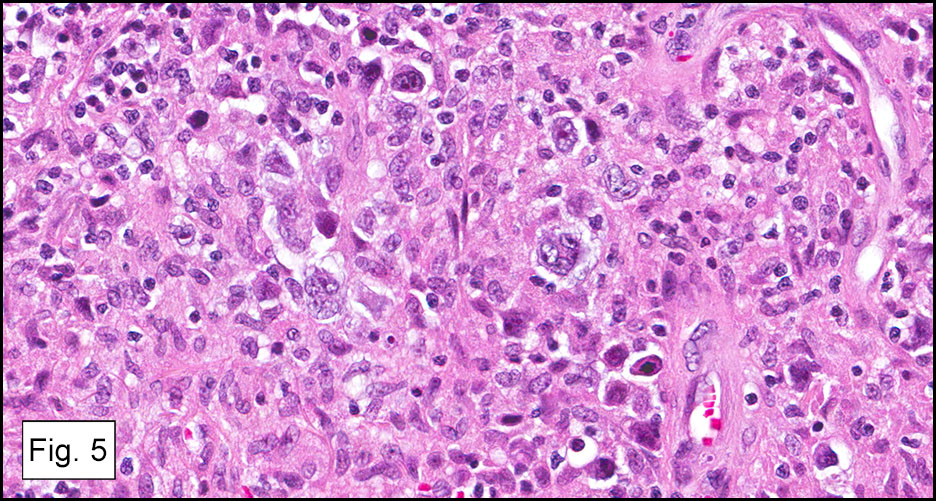
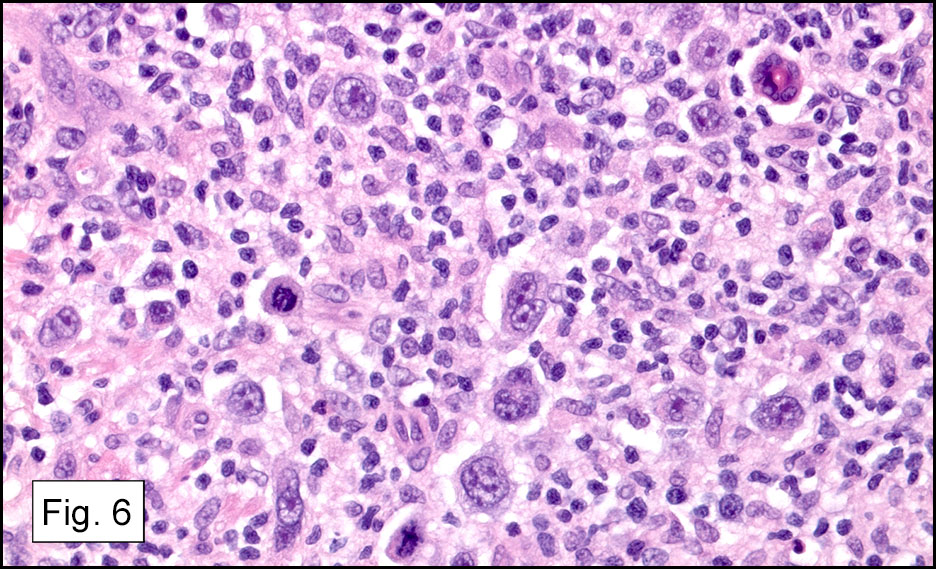
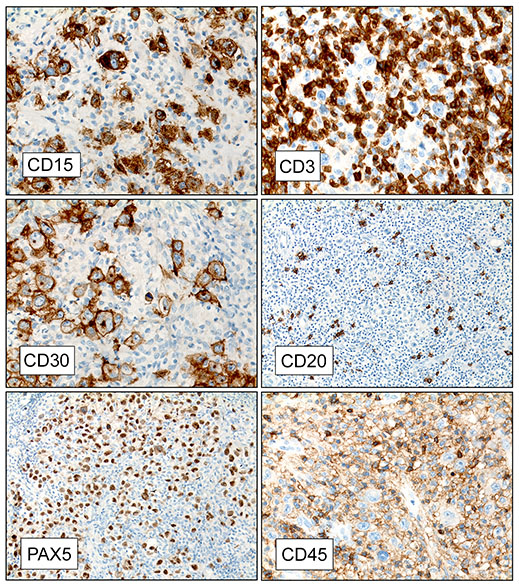
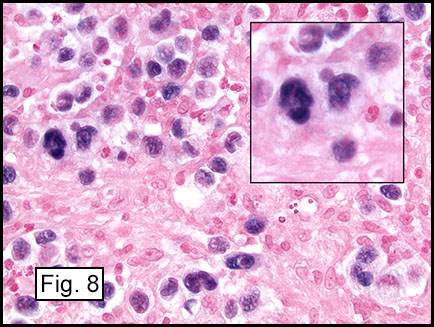
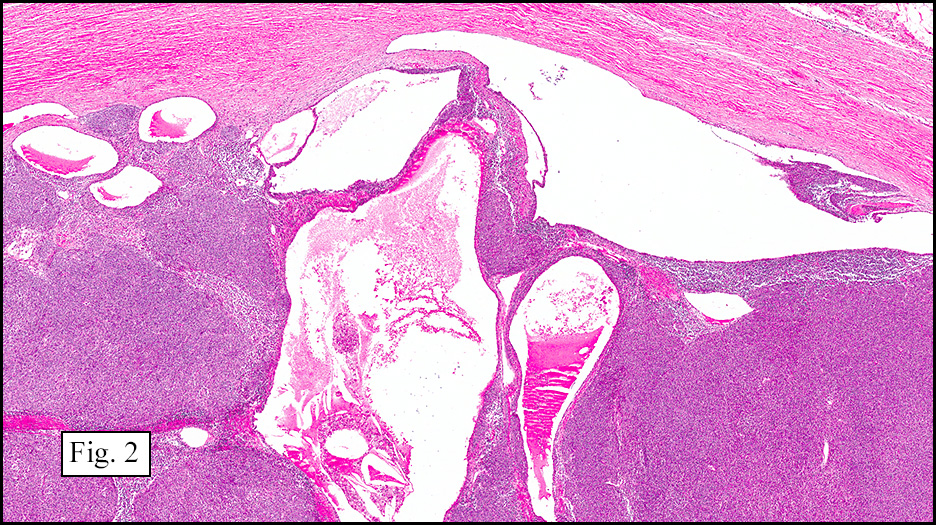
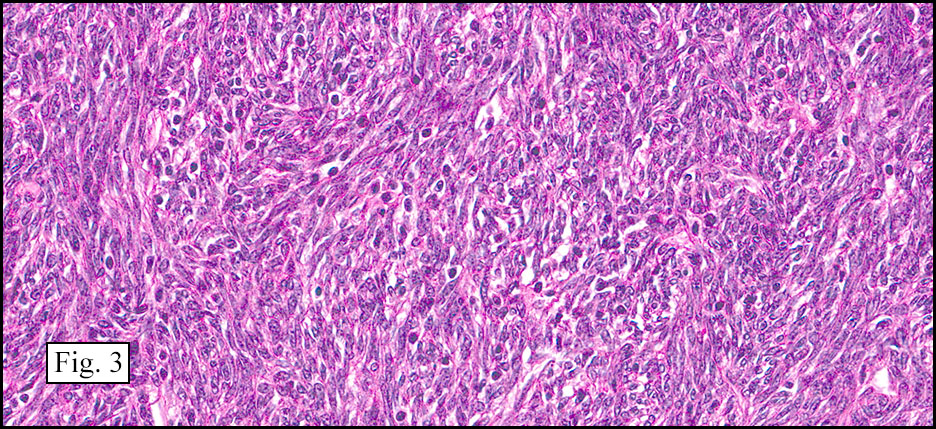
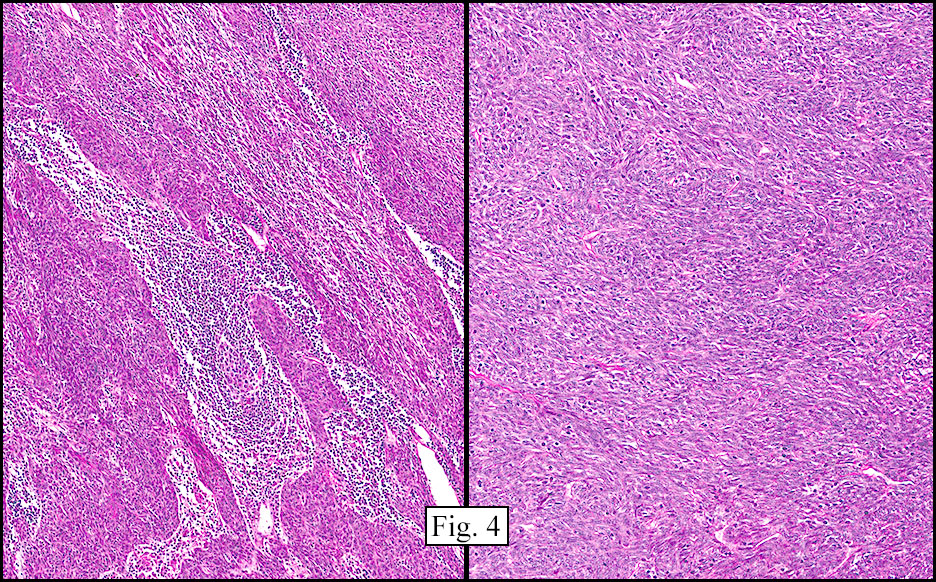
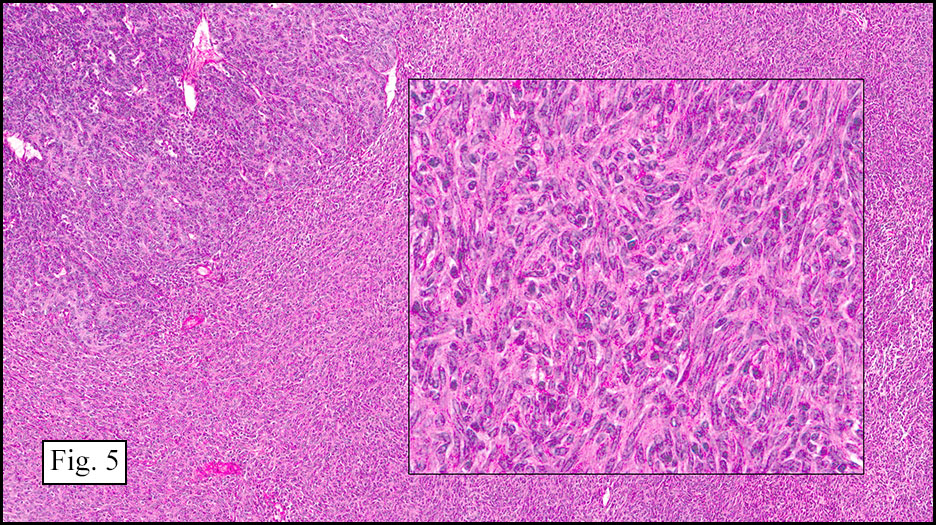
Microscopically, the tumor infiltrated into tendon (Fig. 2). Tumor cells showed a nesting pattern and clear cells with focal pigment (Figs. 3-4). Prominent nucleoli were present (Figs. 5-6). The tumor was positive for a melanoma cocktail (Fig. 7) and S100 (Fig. 8).
Immunohistochemical studies showed the tumor to be positive for S100 and Melanoma cocktail, but were negative for synaptophysin and NFP. FISH was performed and showed 97% tumor cells were positive for fusion transcript of EWSR1(22q12) and ATF1 (12q13.13) loci.
Diagnosis: Clear Cell Sarcoma of Soft Parts
Huina Zhang, M.D., Ph.D., Ravi Raghavan, M.D.
and Donald R. Chase, M.D.
Department of Pathology & Human Anatomy
Loma Linda University Medical center, Loma Linda, California
Discussion: Clear cell sarcoma (CCS) is a rare soft tissue sarcoma with melanocytic differentiation, which is also known as malignant melanoma of soft parts. Although it has certain morphologic similarities to malignant melanoma, CCS is a clinicopathologically and genetically distinct from the conventional malignant melanoma. It mainly affects young adults between the ages of 10 to 40 years with a median age of approximately 30 years. CCS typically presents as a slowly growing, firm, deep-seated nodule in the distal extremities, most often located around the ankle or foot, followed by the knee, wrist, and hands. It is usually associated with tendons, tendon sheaths, or aponeuroses, with rare involvement of the subcutaneous tissue or dermis. CCS usually follows a protracted clinical course, with frequent local recurrences and late metastases to regional lymph nodes, lungs, and bone. The long-term prognosis of CCS is poor and one study showed the survival rates at 5, 10, and 20 years were 67%, 33% and 10%, respectively. Early diagnosis and initial wide excision are essential for local control and a more favorable outcome. Unfavorable prognostic factors include large tumor size (>5 cm), presence of necrosis, early local recurrence, and positive resection margins.
Grossly, CCS is usually a lobulated or multi-nodular, firm, tan-grey mass and the size ranges from 0.4 cm up to 14.5 cm in greatest diameter. It may show infiltration into the tendons and aponeuroses. The cut surface may show focal haemorrhage, necrosis or cystic changes and often is gritty.
Microscopically, CCS is highly infiltrative and is characterized by a nested, bundled or fascicular growth of uniform fusiform or spindled-to-epithelioid cells, separated by prominent fine collagenous framework of various thicknesses. The tumor cells contain abundant clear or pale eosinophilic cytoplasm and centrally located, round to ovoid vesicular nuclei with prominent basophilic nucleoli. Scattered multinucleated giant cells with 10 to 15 peripherally distributed nuclei in a wreathlike pattern are commonly present. In about half to two thirds of cases finely granular melanin pigment may be identified. The stroma may be barely visible, fibrotic or hyalinized. Focal areas of necrosis may be also noted. The mitotic rate is usually low, and pleomorphism is absent.
Immunohistochemical studies of CCS reveal that the tumor cells express antigens associated with melanin synthesis including diffuse immunoreactivity with HMB-45, nuclear and cytoplasmic immunoreactivity to S-100 protein, and reactivity with the microphthalmia transcription factor (MITF). There may be focally reactivity with Melan-A, neuron-specific enolase and vimentin, but the tumor is typically negative for EMA, keratins, and desmin.
Genetically, CCS has a distinct genetic background including a reciprocal chromosome translocation t(12;22)(q13;q12), which can be detected by conventional chromosomal analysis, FISH or reverse transcriptase polymerase chain reaction in 70-90% of CCS. This translocation results in fusion of the 3’ portion of the Ewing sarcoma (EWSR1) oncogene on chromosome 22q12 with the 3’ portion of the activating transcription factor 1 (ATF1) oncogene on chromosome 12q13, giving rise to the EWSR1/ATF1 chimeric transcript. EWSR1/ATF1 functions as a transcriptional regulator that induces the expression of MITF in CCS and plays a crucial role in melanocytic differentiation and growth or survival of tumor cells.
Differential Diagnoses:
• Metastatic melanoma is the most important mimic of CCS in the soft tissue and can be extremely difficult to distinguish from CCS because the significant overlapping features at morphologic, immunophenotypic and ultrastructural levels. The clinical context including a deep-seated infiltrative mass in the distal extremities, no proceeding history of cutaneous, mucosal or ocular tumor, the absence of junctional activity, and the uniform cytomorphology are helpful clues to the diagnosis of CCS, but in some cases detection of the translocation or its gene product is needed.
• Paraganglioma-like dermal melanocytic tumor is a recent-described, rare, benign neoplasm derived from melanocytes. It is primarily a non-pigmented, well-demarcated skin nodule at the extremities, most common in adult females. It is composed of clear to amphophilic epithelioid cells in an organoid or nest-like pattern separated by delicate fibrous strands and blood vessels, which is a reminiscent of those of paraganglioma. The tumor cells may also express S-100 protein, Melan-A, HMB-45, and microphthalmia transcription factor but lacks pancytokeratin or smooth muscle actin. FISH analysis of 5 cases revealed an intact EWS gene locus, supporting absence of the clear cell sarcoma 12;22 translocation.
• Malignant peripheral nerve sheath tumor (MPNST) is usually associated with a large peripheral nerve, and usually has manifestations of neurofibromatosis. It has significant myxoid stroma, long fascicles of spindle cells with slender, tapering or wavy nuclei and indistinct cytoplasm. Although it expresses S-100 protein, MPNST is negative for HMB-45 and Melan-A.
Summary: CCS is an extremely rare soft tissue malignancy with melanocytic differentiation, which is characterized by nests and short fascicles of uniform spindled-to-epithelioid cells with pale, eosinophilic cytoplasm and prominent nucleoli, separated by dense fibrous septa. Immunohistochemically, expression of HMB-45 is usually stronger and more diffuse than S-100 protein. Furthermore, t(12;22) translocation with EWSR1-ATF1 fusion is a typical genetic feature in CCS.
Suggested Reading:
Hornick JL. Practical Soft Tissue Pathology: A Diagnostic Approach. Elsevier.
Dim DC, Cooley LD, Miranda RN. Clear cell sarcoma of tendons and aponeuroses: a review. Arch Pathol Lab Med. 2007, 131(1):152-6.
Deyrup AT1, Althof P, Zhou M, Morgan M, Solomon AR, Bridge JA, Weiss SW. Paraganglioma-like dermal melanocytic tumor: a unique entity distinct from cellular blue nevus, clear cell sarcoma, and cutaneous melanoma. Am J Surg Pathol. 2004, 28(12):1579-86.
Goldblum JR, Folpe AL and Weiss SW. Enzinger & Weiss’s Soft Tissue Tumors: 6th ed. Elsevier.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}