History: An adult woman experienced a painful mass in her back. The mass, excised for symptomatic relief, was in the left paraspinal region at the inferior angle of scapula. Clinically, it was 9 x 4 cm, soft and movable. It had been gradually enlarging for about 3 years.
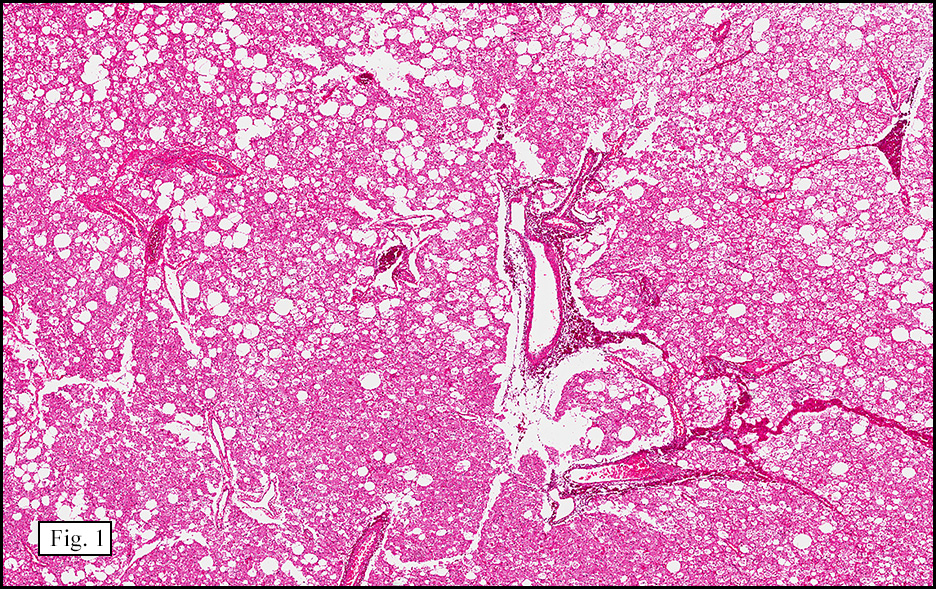
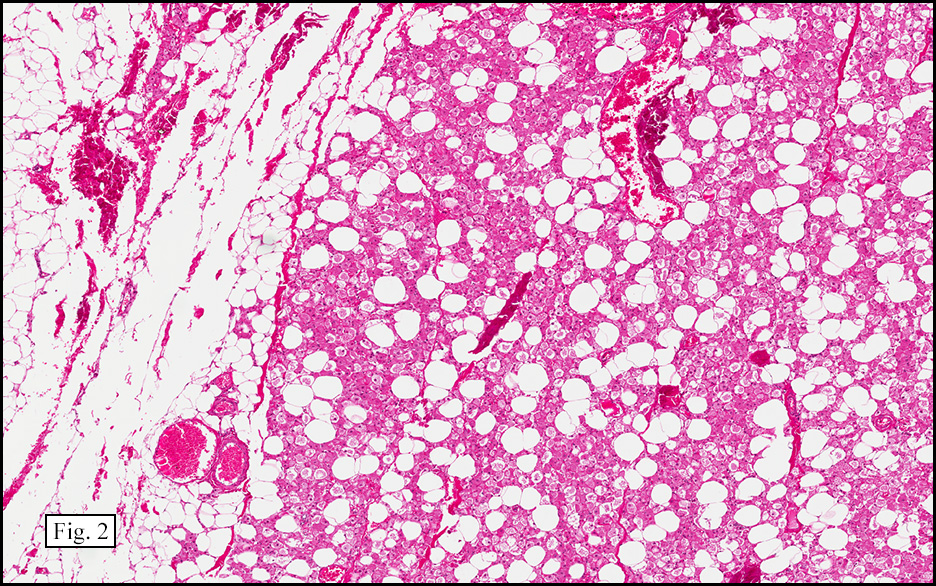
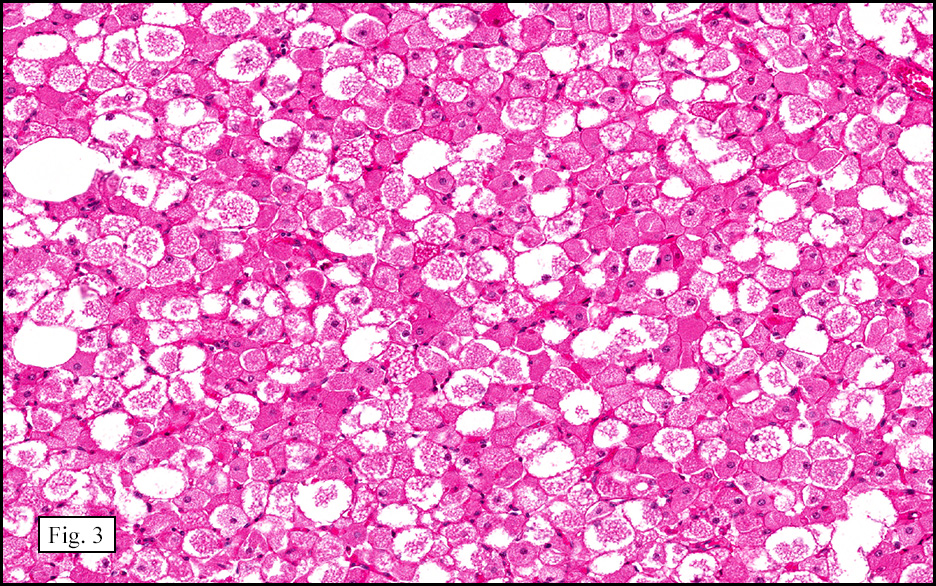
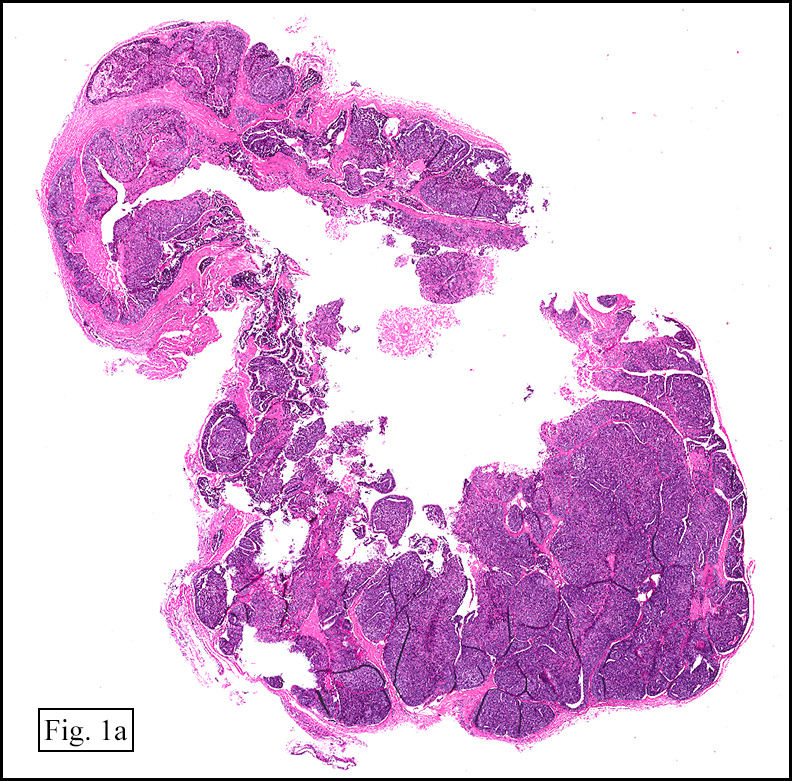
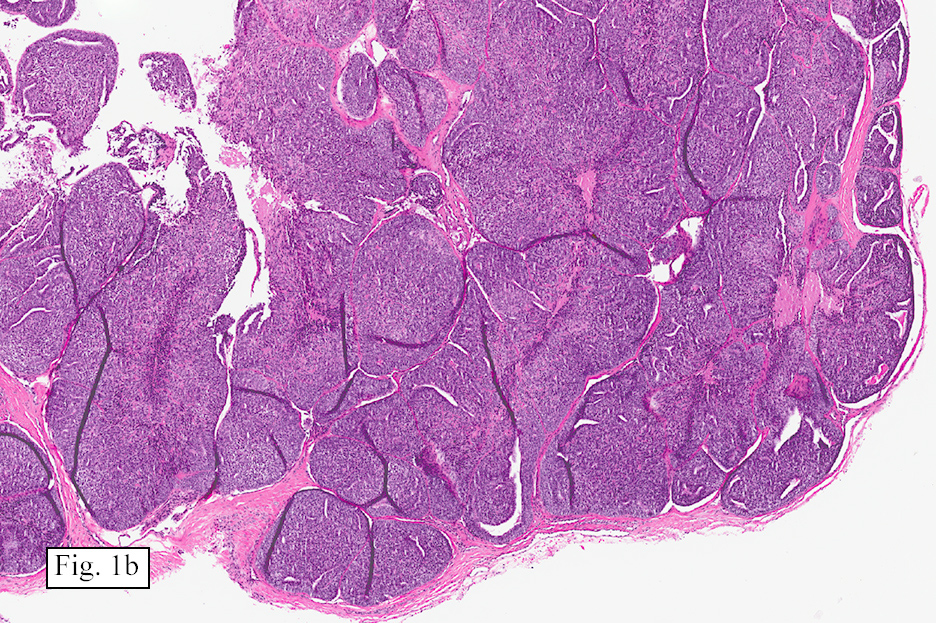
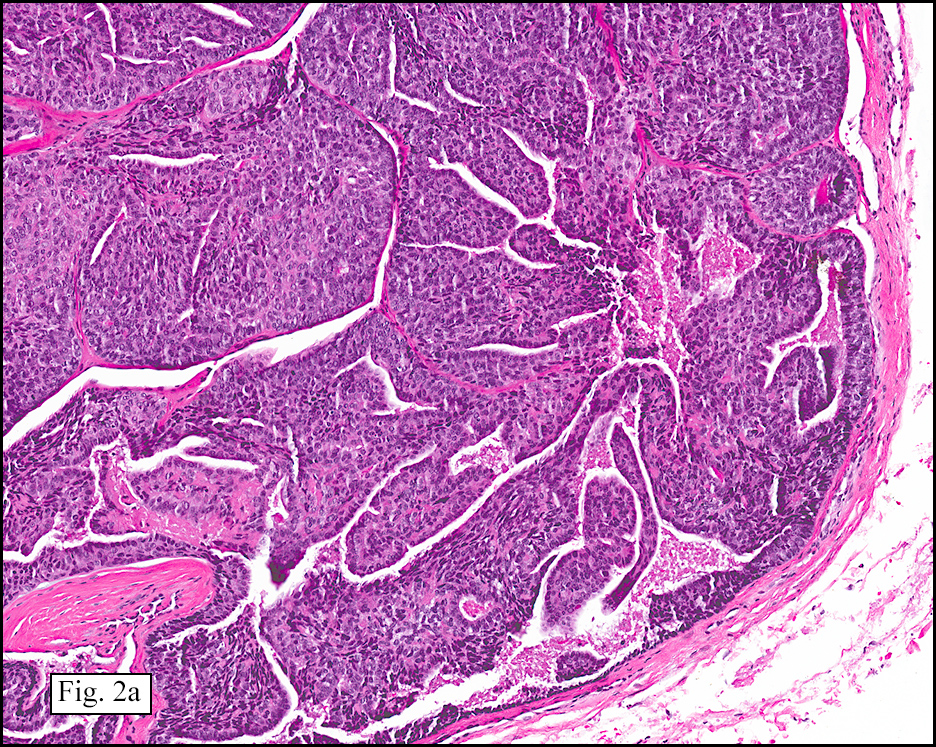
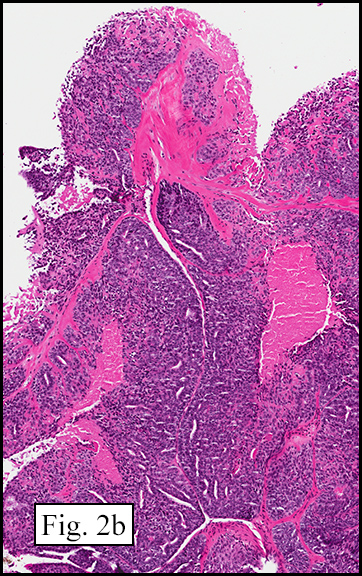
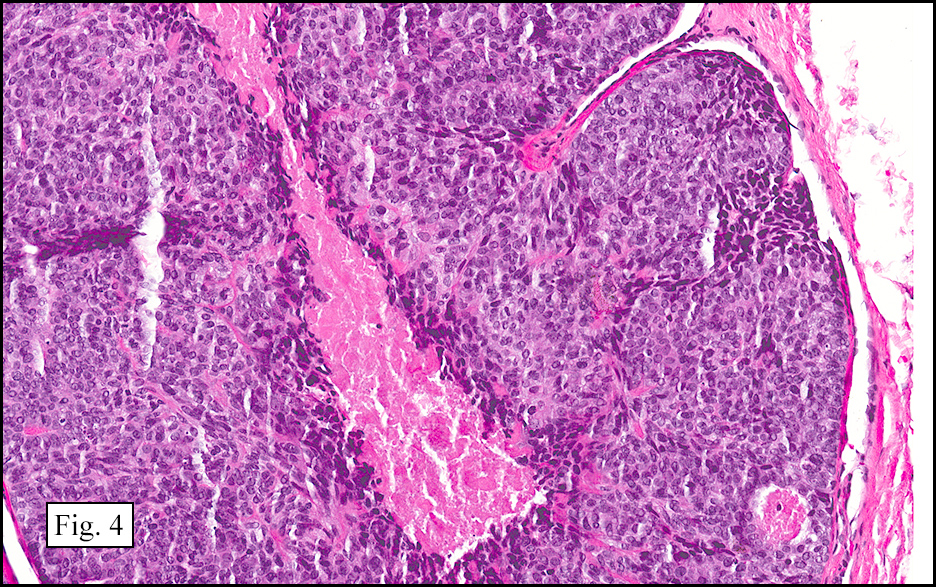
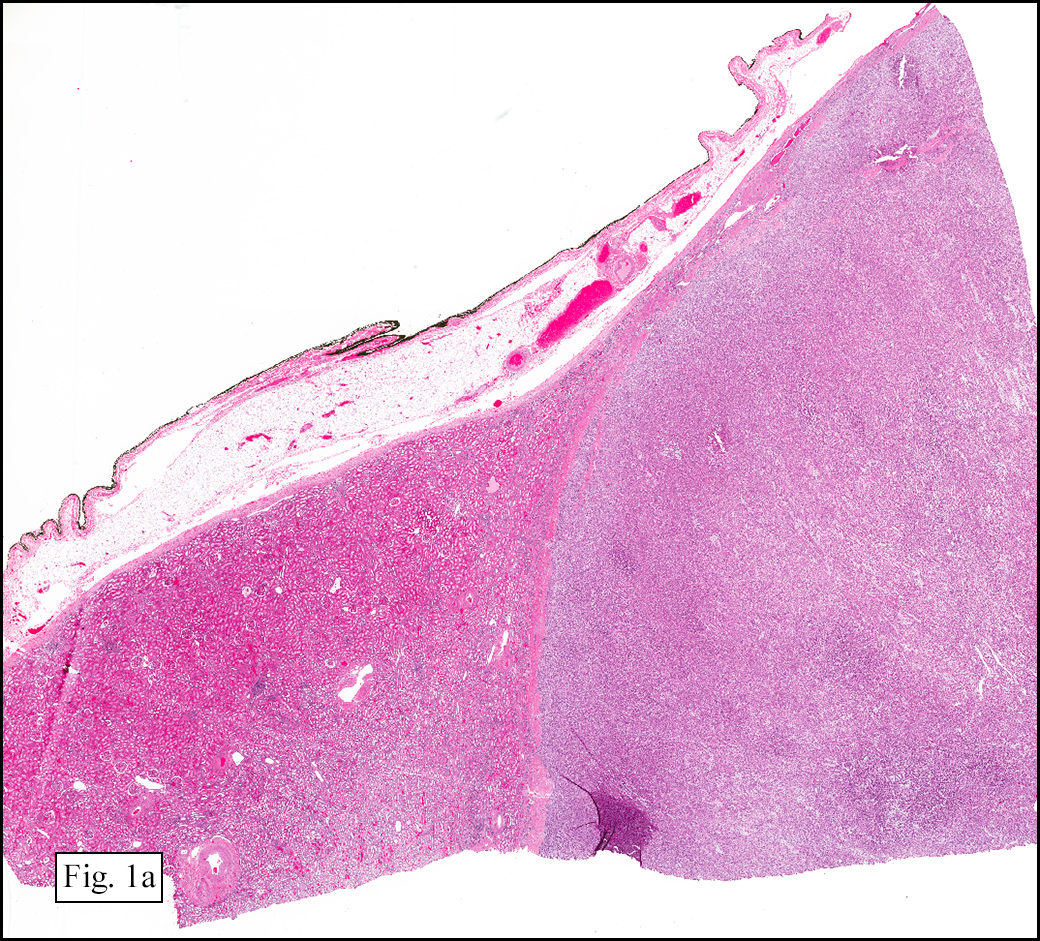
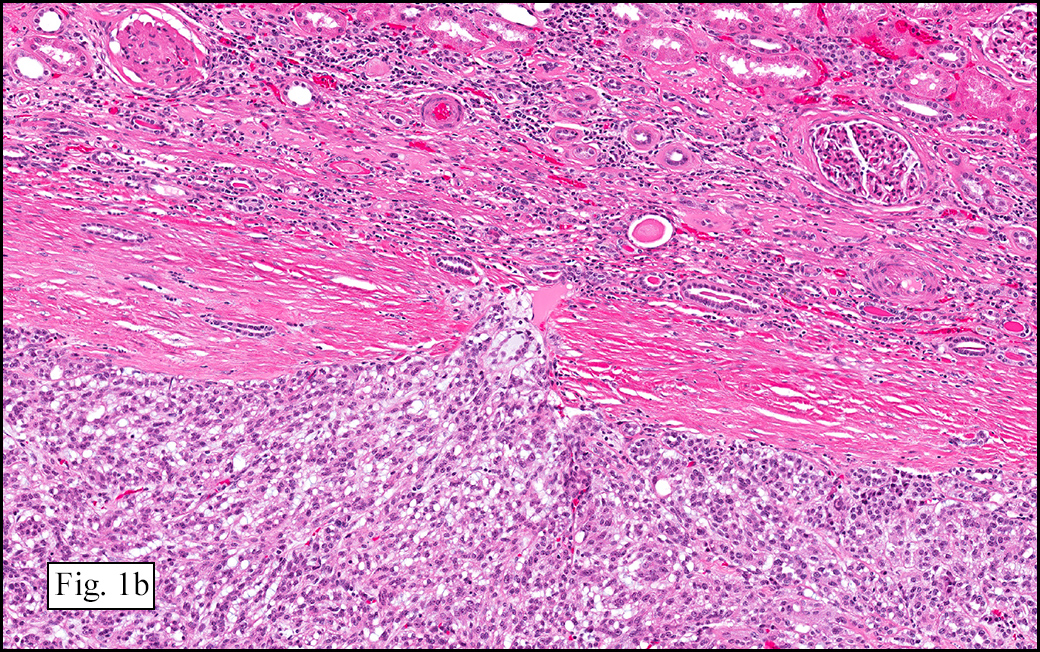
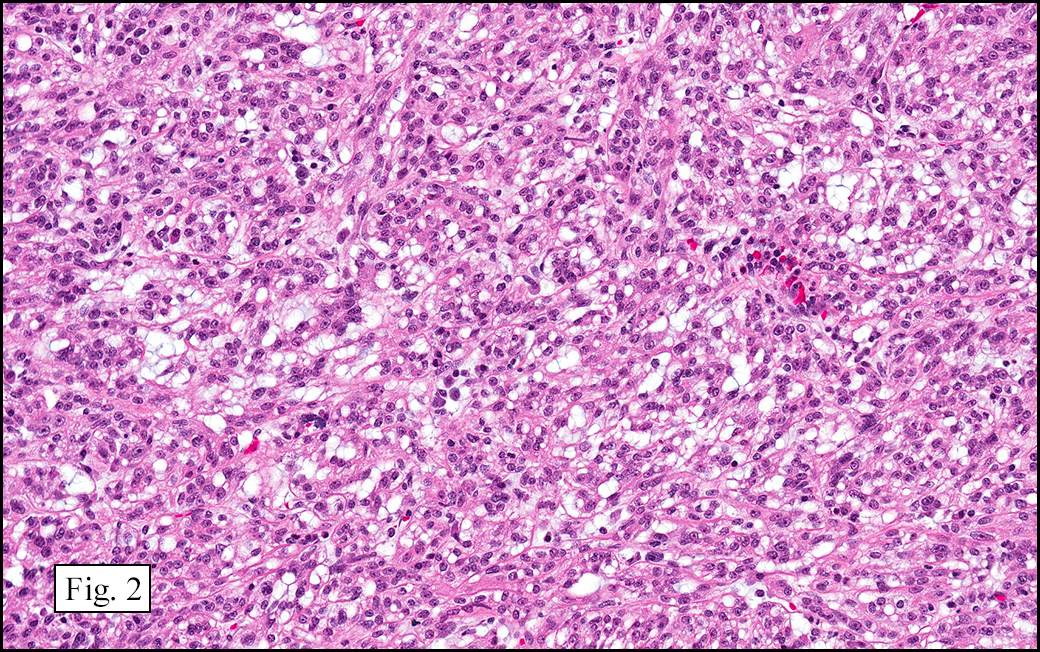
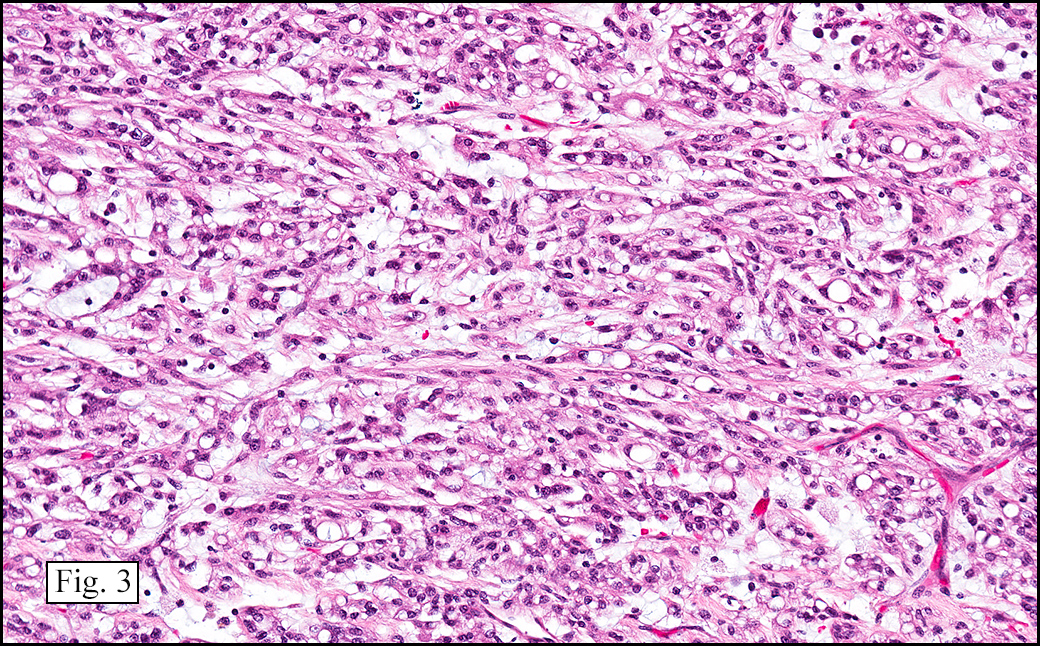
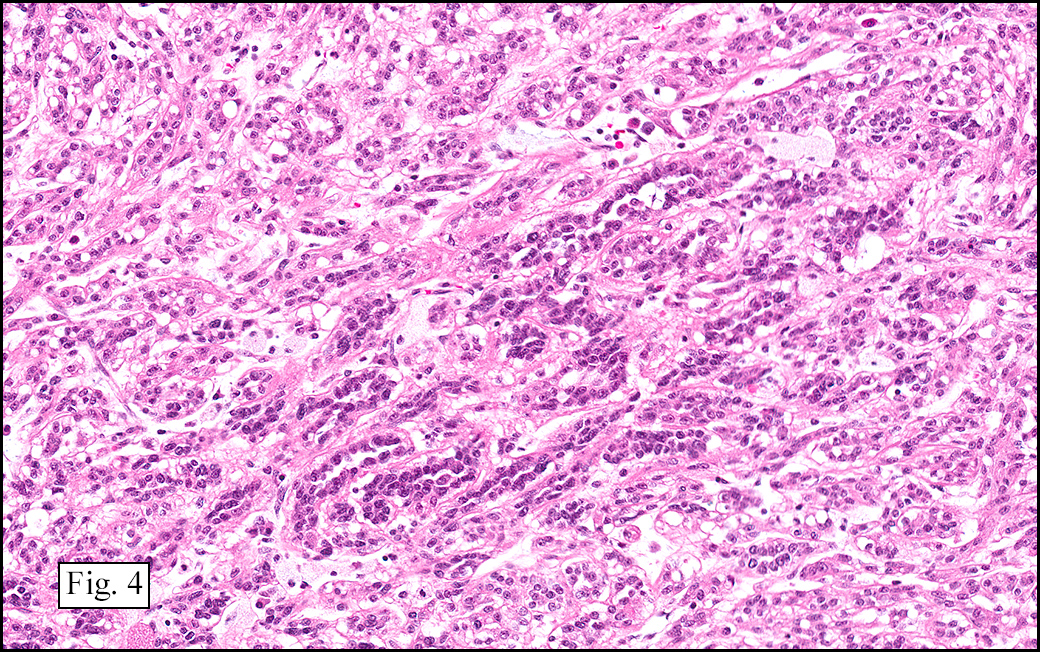
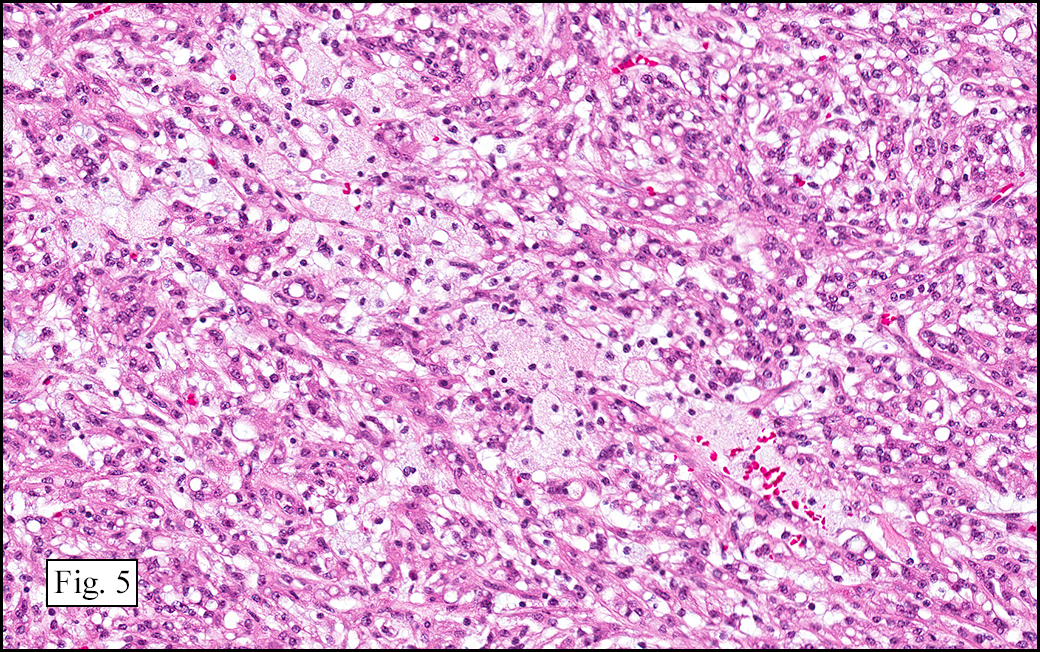
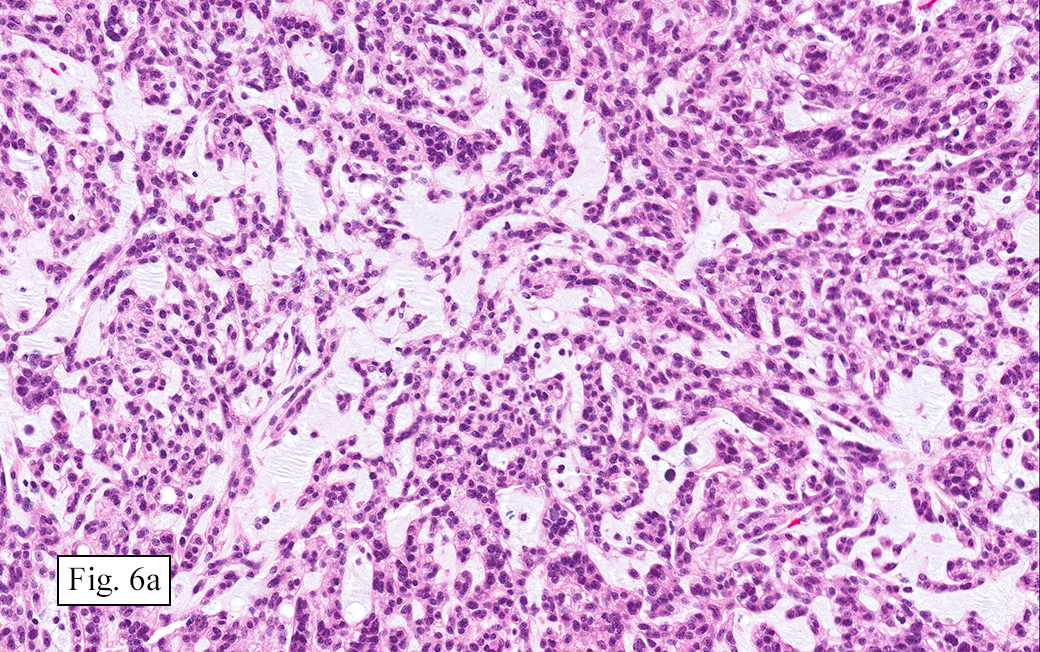
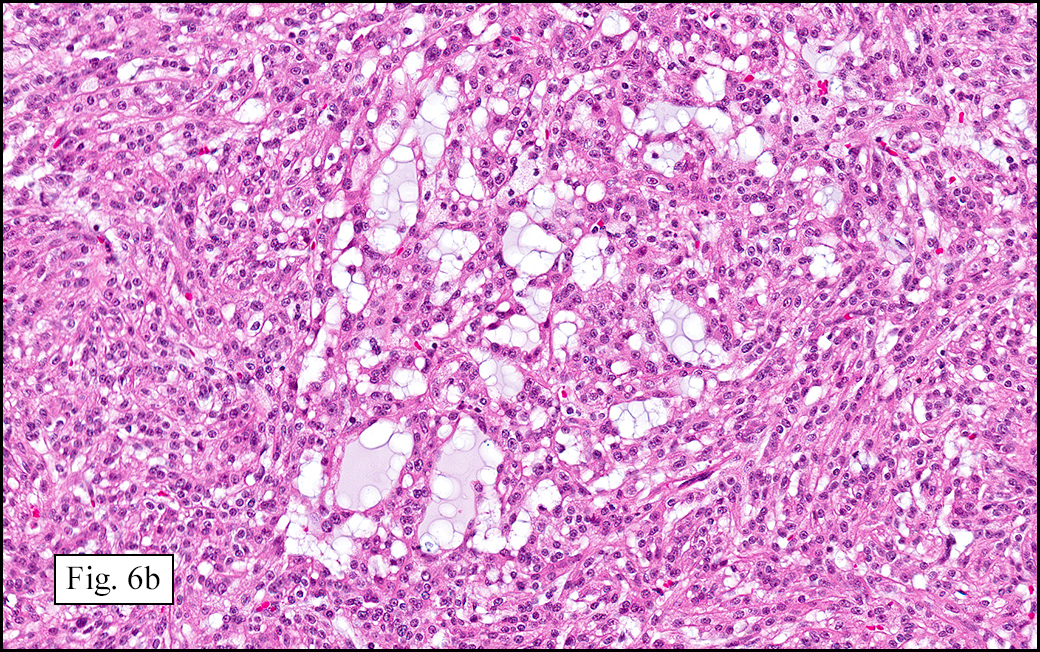
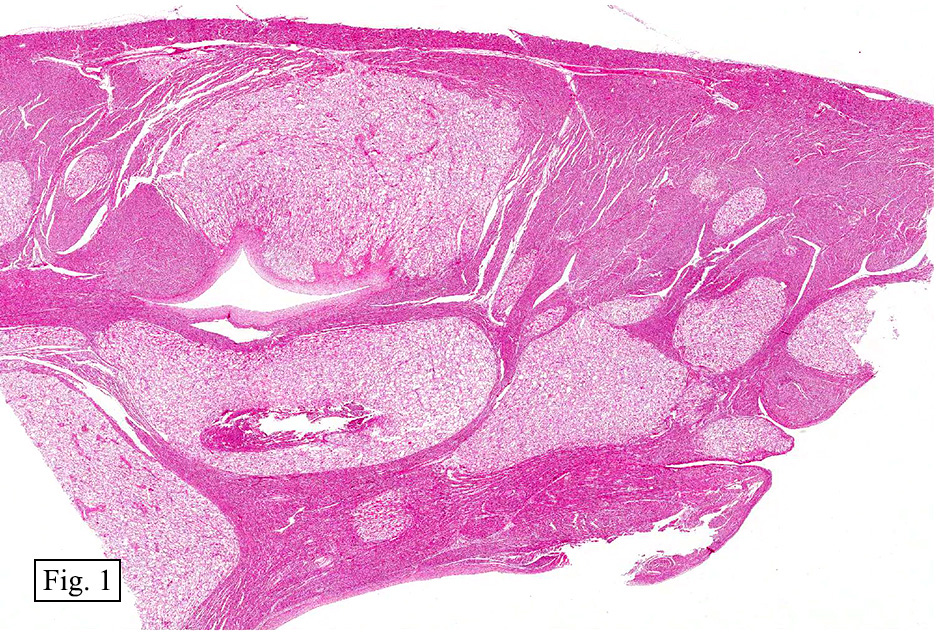
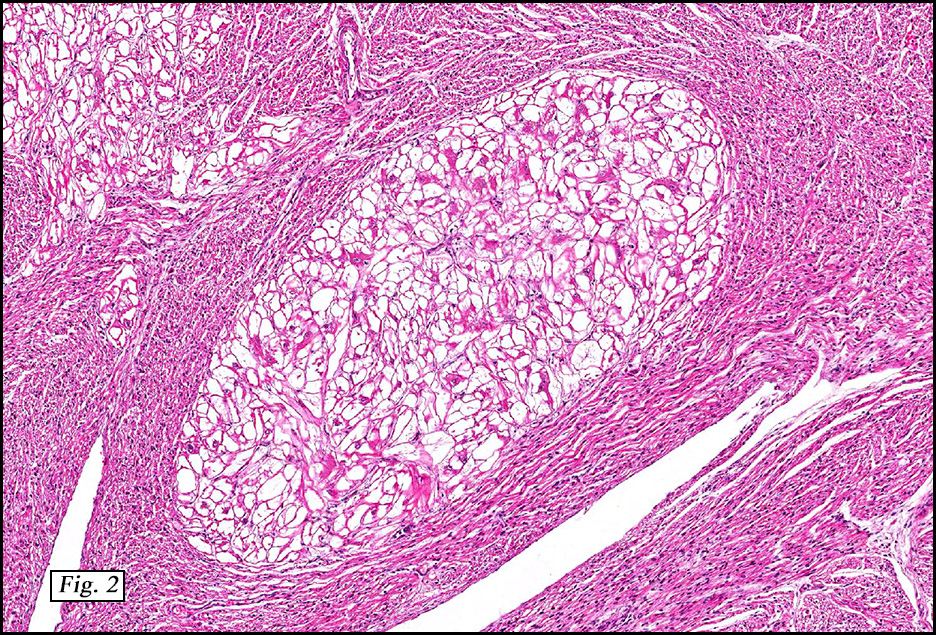
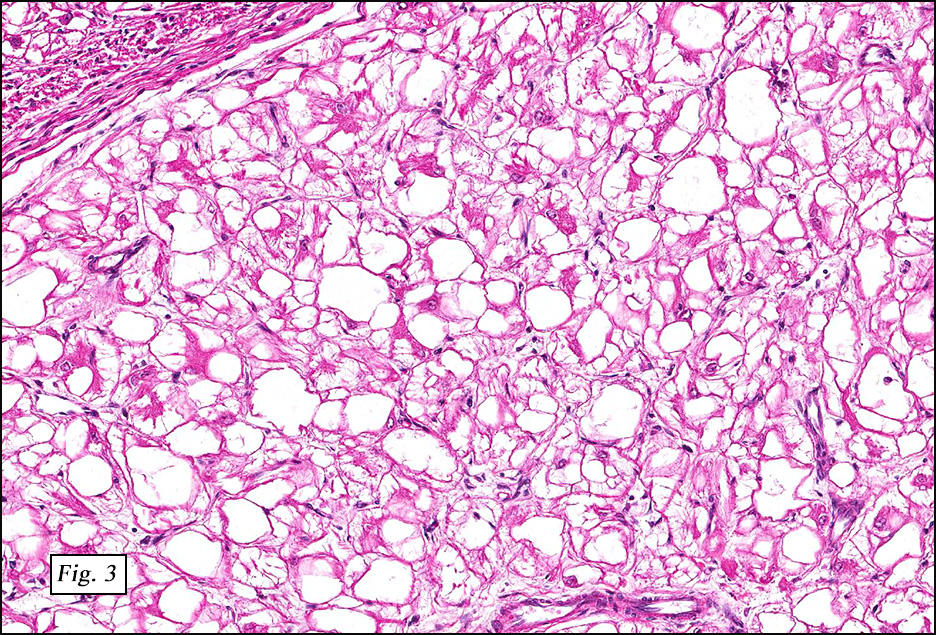
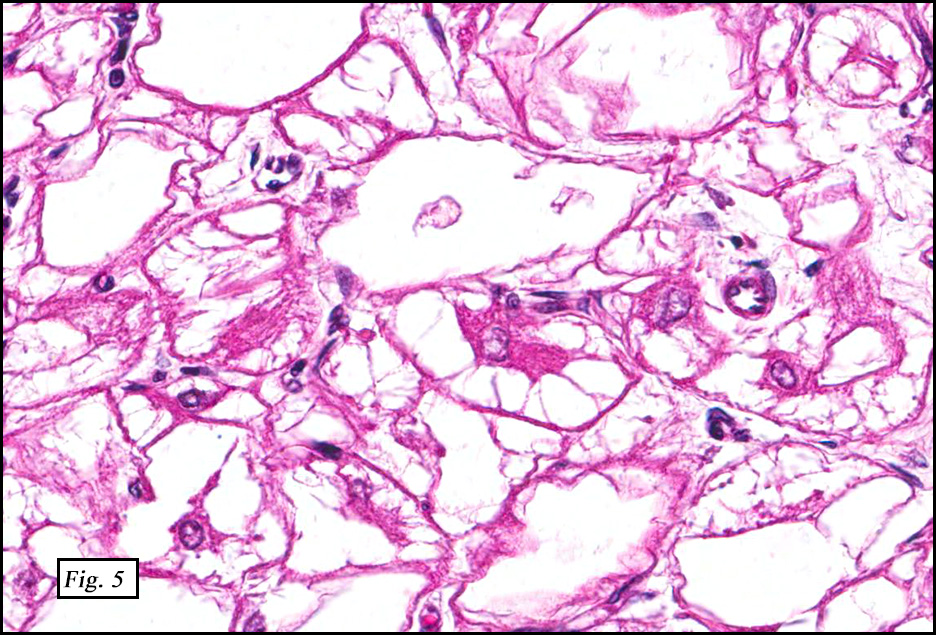
The excised specimen showed a 6 x 5 x 2.5 cm tumor that was composed of tan-brown fibrous tissue. Microscopically, it consisted of sheets of eosinophilic cells with increased vascularity admixed with mature adipose cells (Figs. 1 and 2). Higher power demonstrated round to polygonal, uniform cells with variable granular, eosinophilic, and multivacuolated cytoplasm and central, round nuclei. Neither atypia nor mitotic figures were seen (Fig. 3).
Diagnosis: Hibernoma, Back
Tae Hun Kim, MSIV and Donald R. Chase, M.D.
Department of Pathology & Human Anatomy
Loma Linda University Medical Center,
Loma Linda, California
Discussion: Hibernoma is a rare, benign adipocytic tumor of brown fat, named as such by Gery in 1914 because he felt that the fat resembled that which was found in hibernating animals. Other terms including lipoma of immature adipose tissue, lipoma of embryonic fat, and fetal lipoma have also been used.
Hibernoma arises predominantly in young adults with a peak incidence during the third decade of life but occurrences may span ages 2 to 75 years (mean age of 38). But in general they tend to occur in a younger population than patients with lipomas. Curiously, only 9 of 170 Armed forces Institute of Pathology (AFIP) cases occurred before 18 years of age. The location of these tumors has been thought to be in sites where brown fat persists beyond fetal life, such as neck, back, and retroperitoneum. But the AFIP study demonstrated that almost one third of their cases occurred in the thigh and other sites that lack vestigial brown fat. These observations suggest an altered differentiation pathway towards brown fat rather than a true origin from native brown fat. Like lipomas, hibernomas present as slow-growing, painless, movable tumors that usually arise in the subcutis. Up to 10% can be intramuscular. Because hibernomas usually range from 5-15 cm in diameter, symptoms related to compression of adjacent structures are relatively common.
Ultrasonography demonstrates a well-circumscribed hyperechoic lesion with increased vascularity. On CT, the signal intensity is between that of skeletal muscle and subcutaneous fat. Intravenous contrast administration enhances hibernomas and demonstrates intratumoral septations and vessels. On MRI, signal intensity is less than that of subcutaneous fat on T1-weighted imaging and is increased on T2-weighted imaging. Fat suppression techniques often decreases the signal intensity but is dependent on the fat composition.
Grossly, hibernomas are well-defined, soft tissue masses that are tan to deep red brown due to increased vascularity and high concentration of mitochondrial cytochrome pigments.
Four variants have been described depending upon the amount of regular fat, nature of stroma, and presence of spindle cells:
• Typical (82% of 170 AFIP cases),
• Myxoid (8%),
• Lipoma-like (7%), and
• Spindle cell (2%).
The typical variant demonstrates a distinct lobular pattern consisting of uniform round to oval multivacuolated cells with granular, eosinophilic cytoplasm and central nuclei without atypia and variable amount of white adipose cells.
The lipoma-like variant has a predominance of white adipose cells with scattered hibernoma cells.
The myxoid variant shows hibernoma cells/brown fat in a background of virtually acellular myxoid stroma.
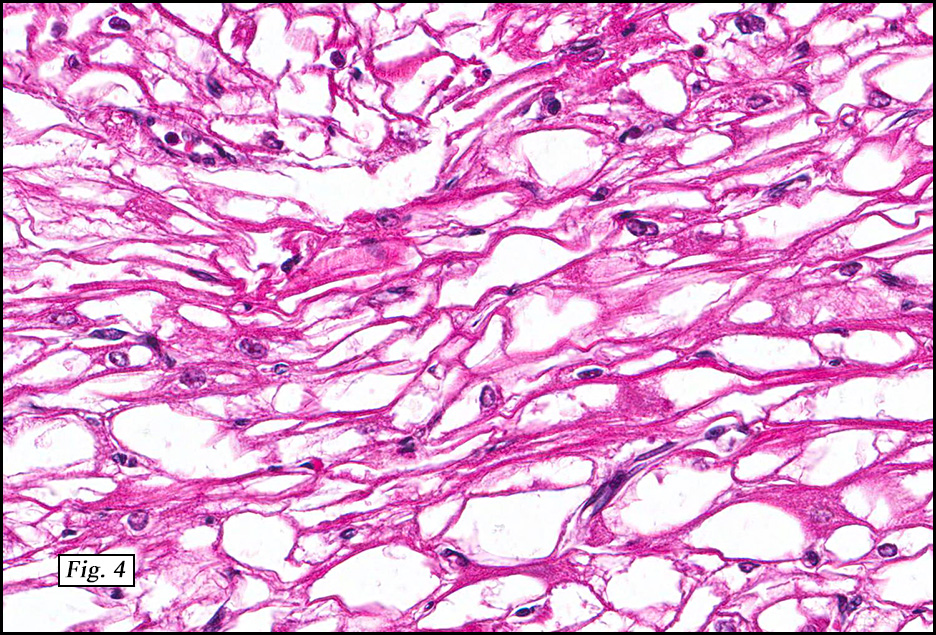
The spindle cell variant is composed of bland spindle cells with multivacuolated hibernoma cells, a hybrid between spindle cell lipoma and hibernoma. This variant often occurs in the neck or scalp. It may have CD34 positivity, while other hibernoma variants are negative.
On electron microscopy, hibernomas are characterized by numerous polymorphous mitochondria.
Immunohistochemically, hibernomas are strongly positive for S-100. They are also positive for Oil Red O and Sudan Black. The karyotypes of hibernomas may be pseudodiploid. Structural rearrangements of 11q13-21 with no consistent chromosome partner are most characteristic. Homozygous deletions of multiple endocrine neoplasia I (MEN1) on 11q13.1 and rearrangements near the GARP gene on 11q13.5 have been implicated in hibernoma pathogenesis.
The differential diagnosis includes:
• Lipomas occur in older adults compared to hibernomas. Lipomas are less vascular and tend to be univacuolated.
• Rhabdomyomas have similar eosinophilic cytoplasm that contains glycogen but lacks lipid vacuoles. Crystals and cross-striations are observed. A desmin stain is usually positive in rhabdomyoma but is negative in hibernoma.
• Granular cell tumors are S-100 positive but lack vacuolated cytoplasm and cytoplasmic lipid vacuoles.
• Well-differentiated liposarcoma (WDLS) lacks brown fat, but may show lipoblasts which can mimic hibernoma cells.
• Myxoid liposarcoma can be confused with the myxoid hibernoma variant as both have multivacuolated cells in a myxoid stroma, but myxoid liposarcoma have mild nuclear atypia, signet ring lipoblasts, and arborizing vessels not found in hibernoma.
Hibernomas regardless of variant typing are treated by complete surgical excision with rare recurrence. Neither metastasis nor malignant transformation have been reported.
Suggested Reading:
Dagher W, et al, Hibernoma presenting as an asymptomatic neck mass, Am J Otolaryngol–Head and Neck Med and Surg (2013), http://dx.doi.org/10.1016/j.amjoto.2013.07.006
Fletcher CDM, Unni KK, Mertens F. World Health Organization Classification of Tumours: Pathology and Genetics of Tumors of Soft Tissue and Bone: Lyon, IARCPress, 2002: 33-34.
Furlong MA, Fanburg-Smith JC, Miettinen M. The morphologic spectrum of hibernoma: a clinicopathologic study of 170 cases. Am J Surg Pathol. 2001;25(6):809 – 14.
Kempson RL, Fletcher CDM, Evans HL, Hendrickson MR, Sibley RK. Atlas of Tumor Pathology: Tumors of the Soft Tissue, 3rd Series Fascicle 30: Washington, D.C., Armed Forces Institute of Pathology, 1998: 197-198.
Liu W, Bui MM, Cheong D. Hibernoma: comparing imaging appearance with more commonly encountered benign or low-grade lipomatous neoplasms. Skeletal Radiol 2013; 42:1073-1078.
Vassos N, et al, Deep-seated huge hibernoma of soft tissue: a rare differential diagnosis of atypical lipomatous tumor/well differentiated liposarcoma. Int J Clin Exp Pathol 2013;6(10):2178-2184.
Weiss S, Goldblum J. Enzinger & Weiss’ Soft Tissue Tumors, 5th ed: Philadelphia, Mosby/Elsevier Inc, 2008; 466-470.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}