History: A 74-year-old male presented with worsening gastric pain, and a prior history of Helicobacter pylori infection.
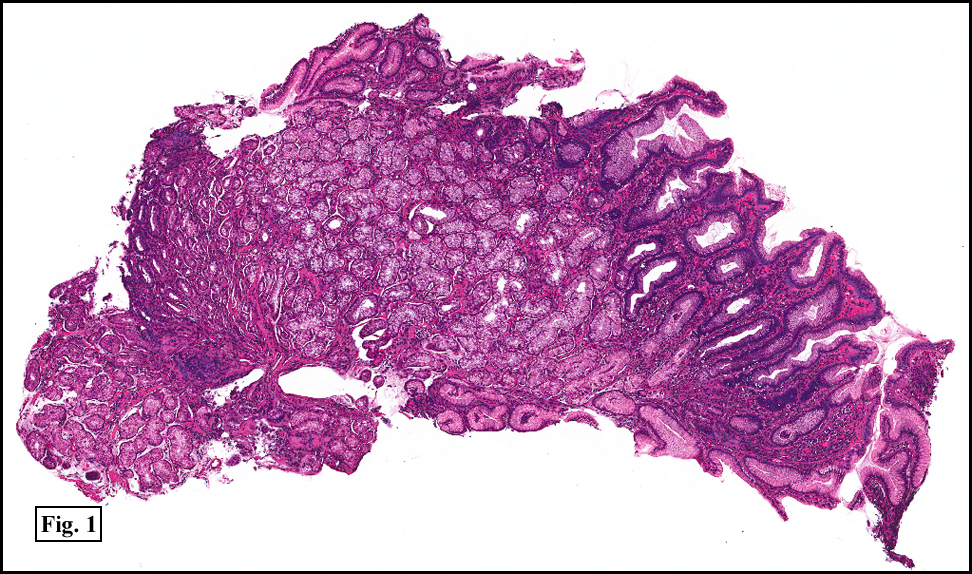
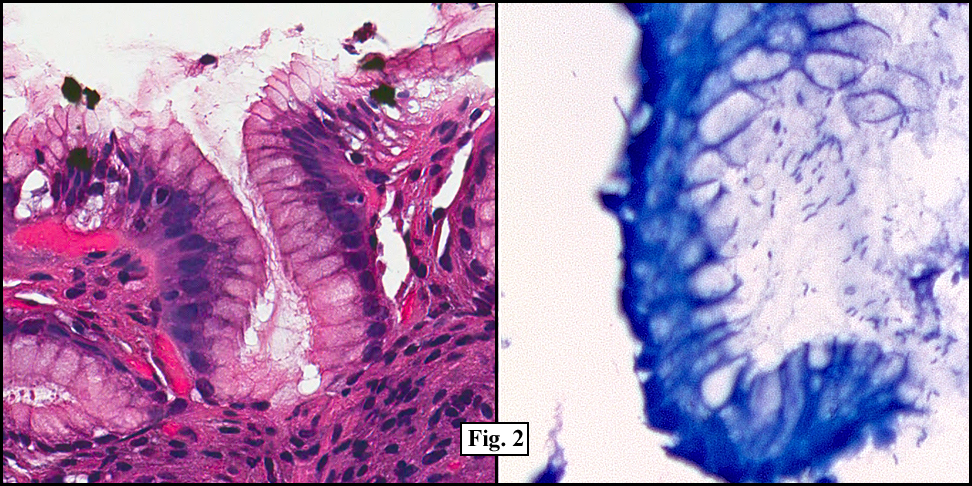
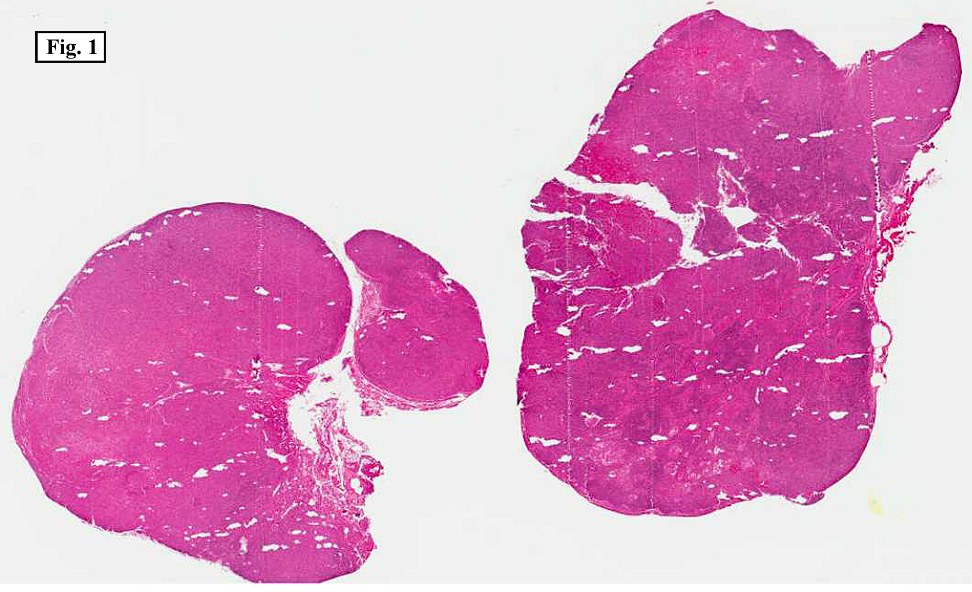
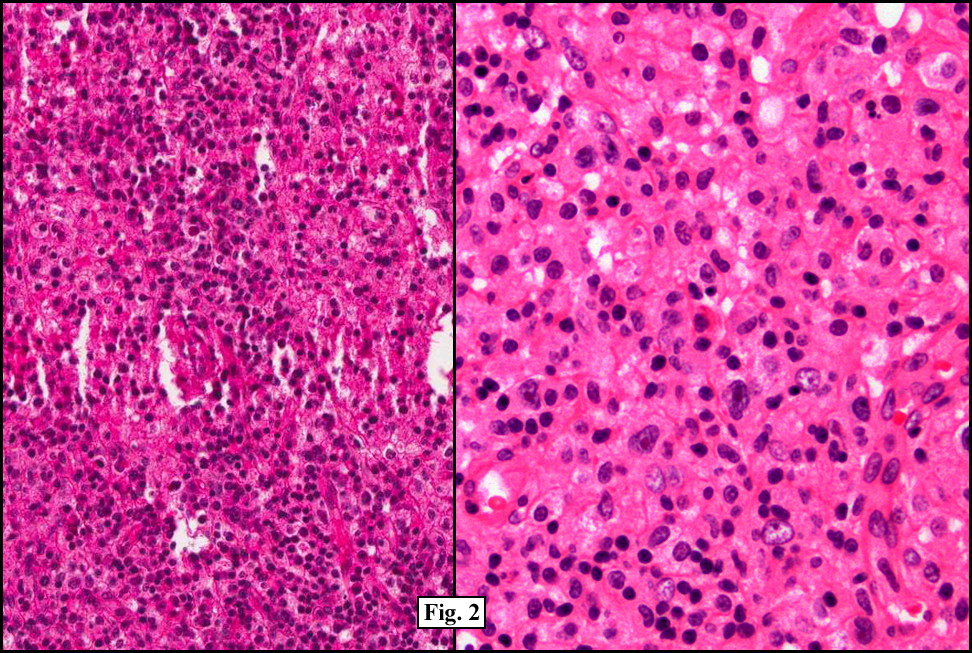
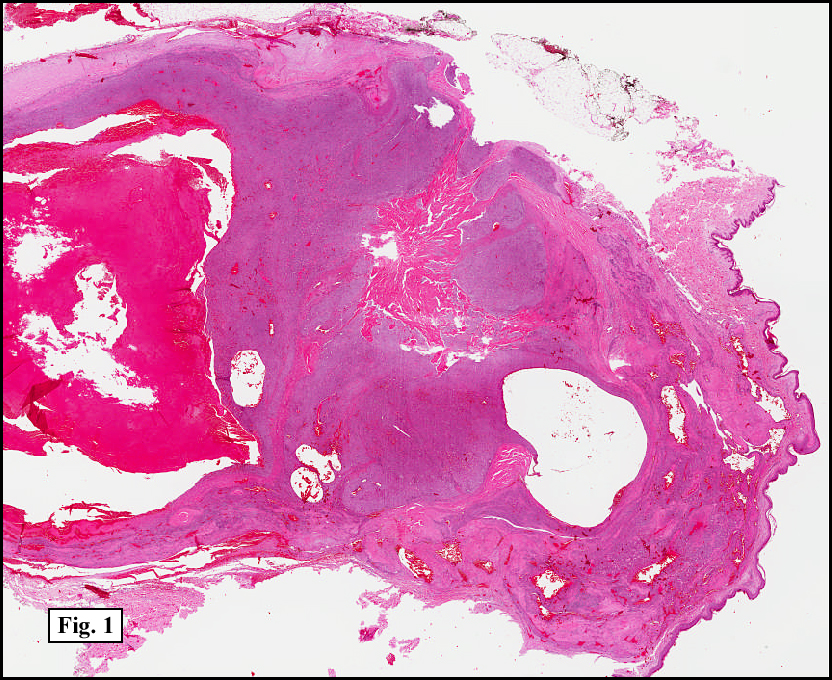
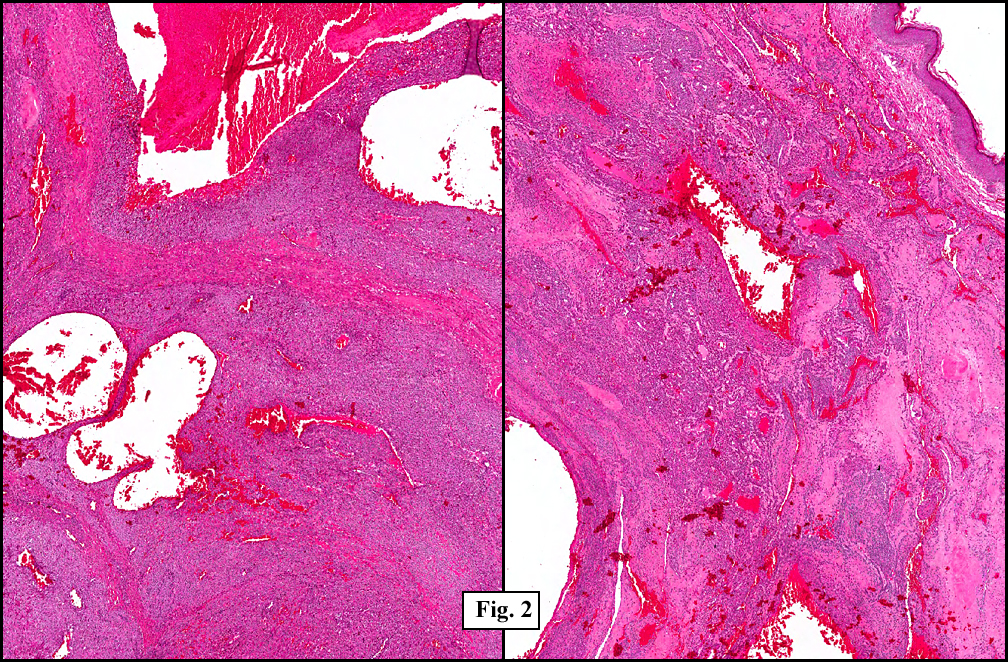
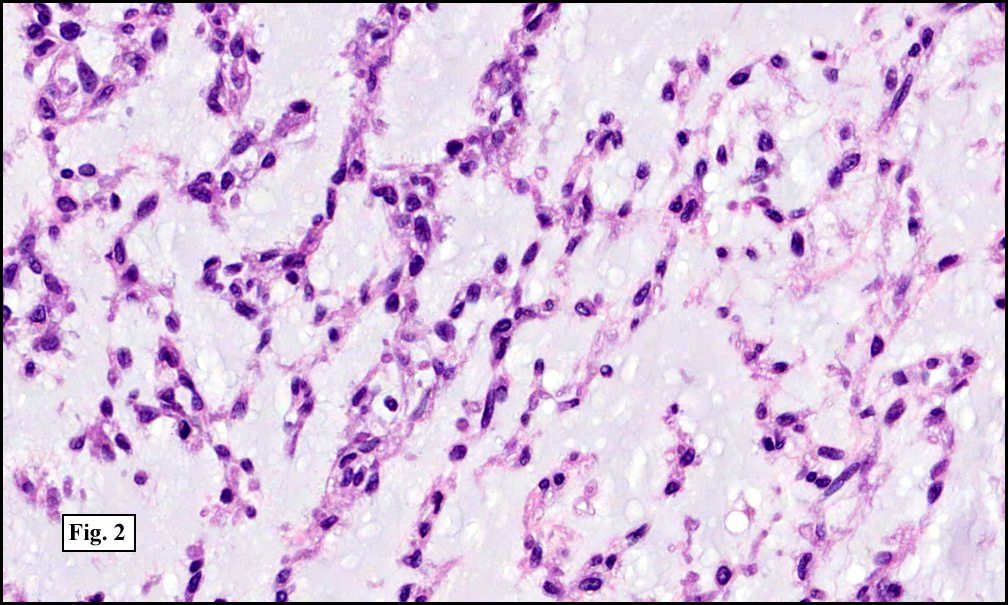
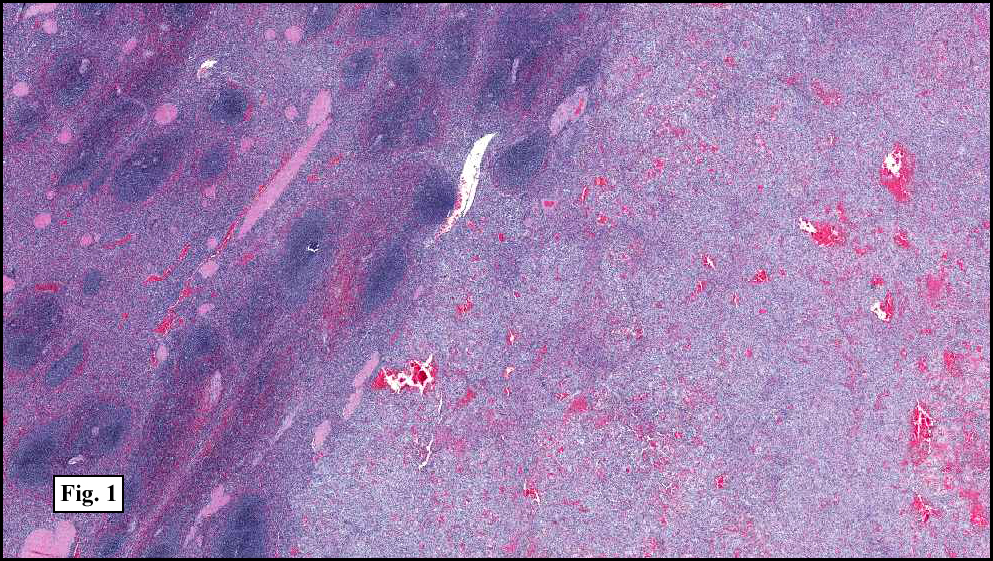
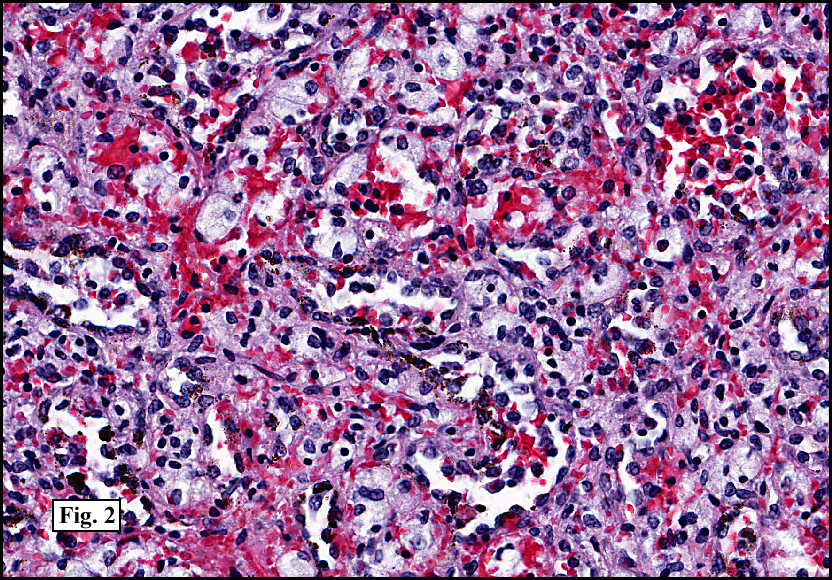
Two years earlier he had an endoscopic biopsy of the gastric body and antrum which revealed moderate acute and chronic inflammation consisting of a superficial mucosal infiltrate of lymphocytes, plasma cells, neutrophils and scattered eosinophils (Fig. 1). A Giemsa stain showed numerous curved bacterial rods present within the gastric crypts and on the surface of the glands (Fig. 2). He was treated with the standard triple therapy (omeprazole, metronidazole and clarithromycin) with resolution of dyspepsia.
{kind=link}
{kind=link}
He now presented with a 4 to 5 month history of abdominal pain and anemia. Endoscopic studies revealed a large fundic mass. A biopsy was performed.
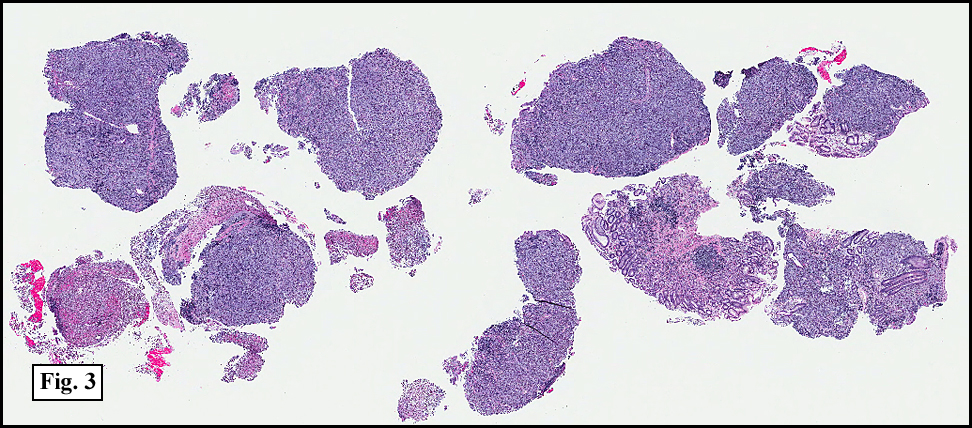
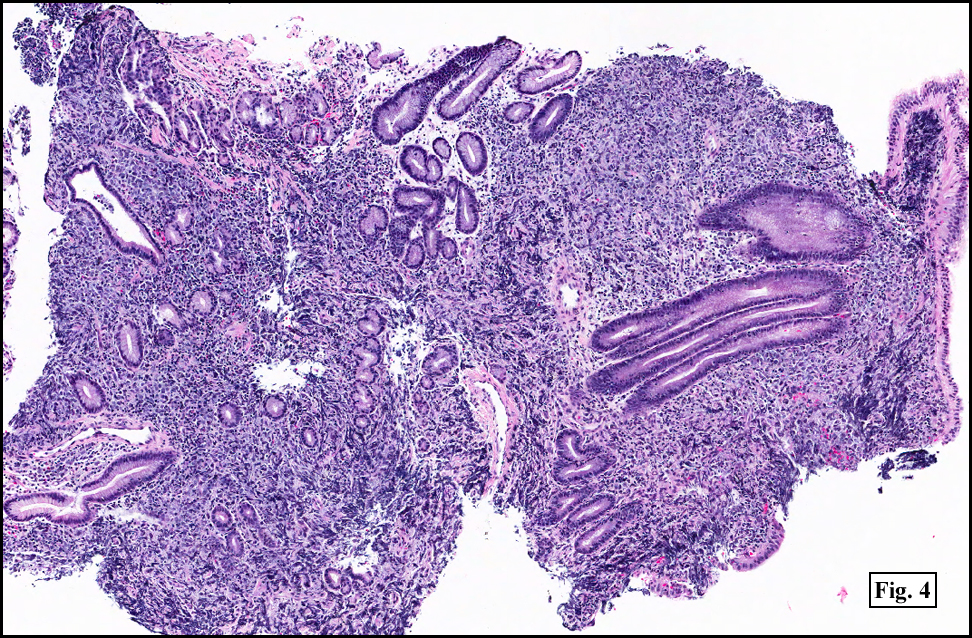
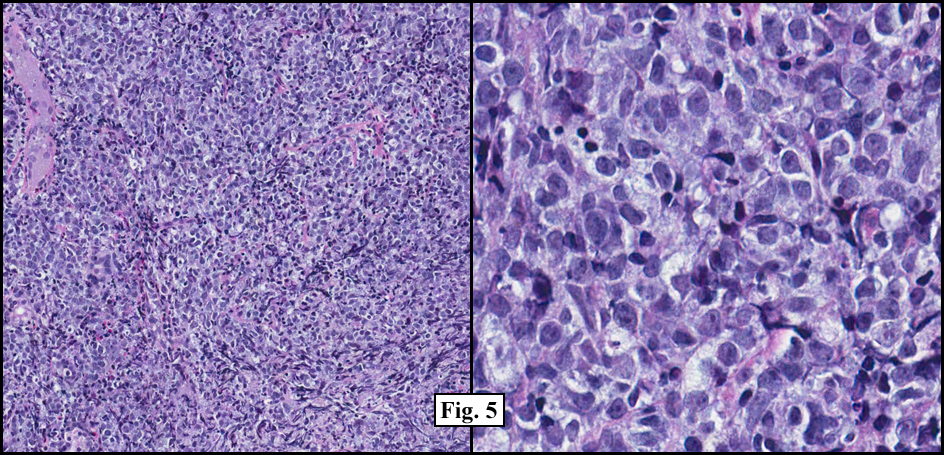
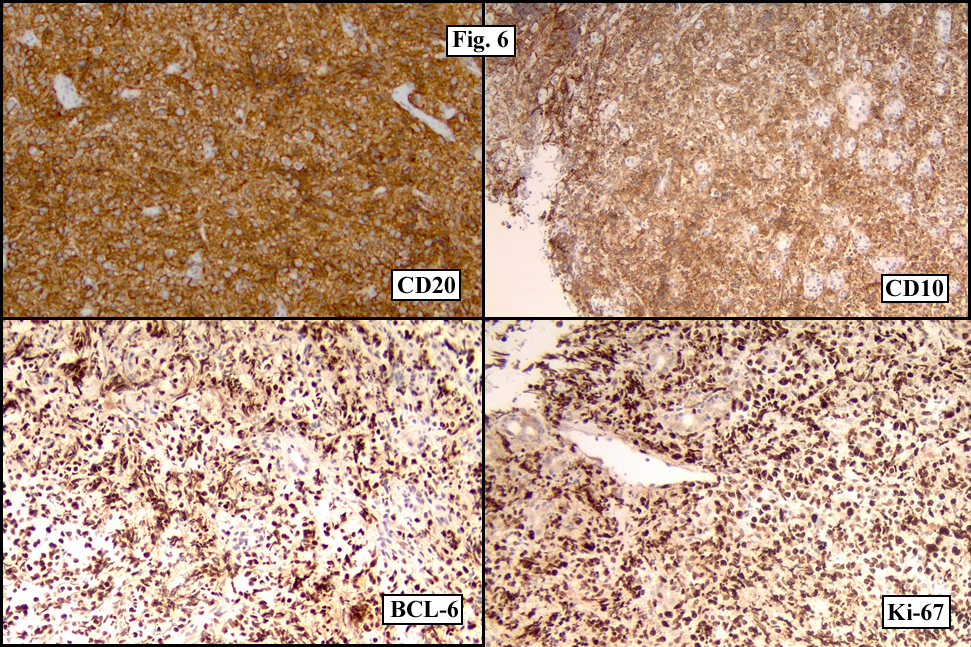
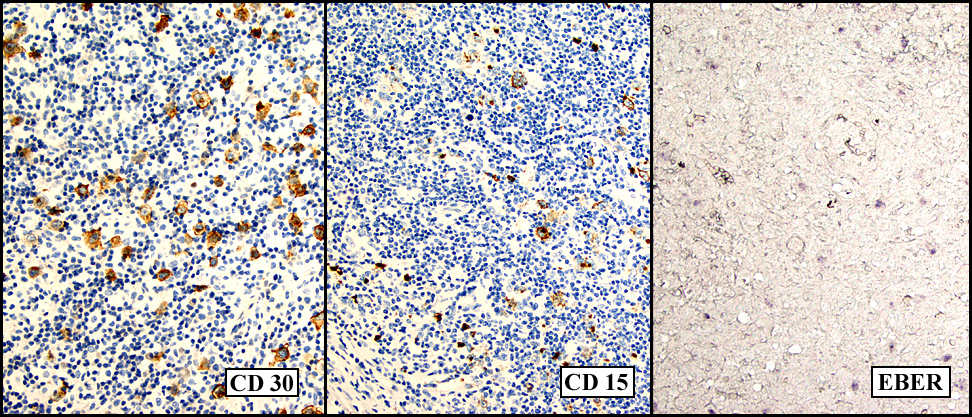
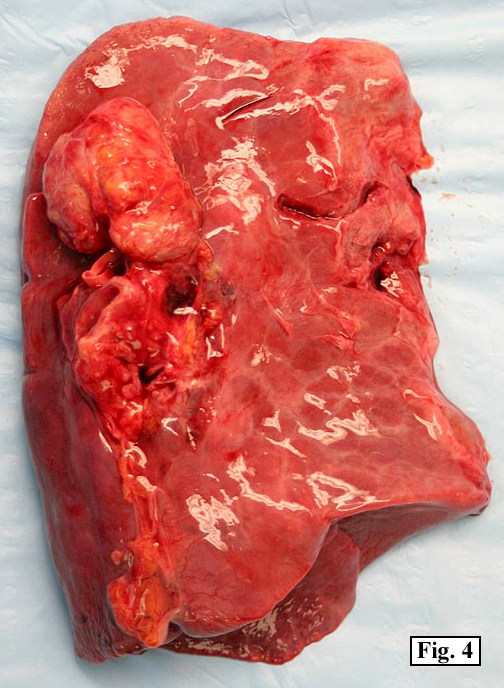
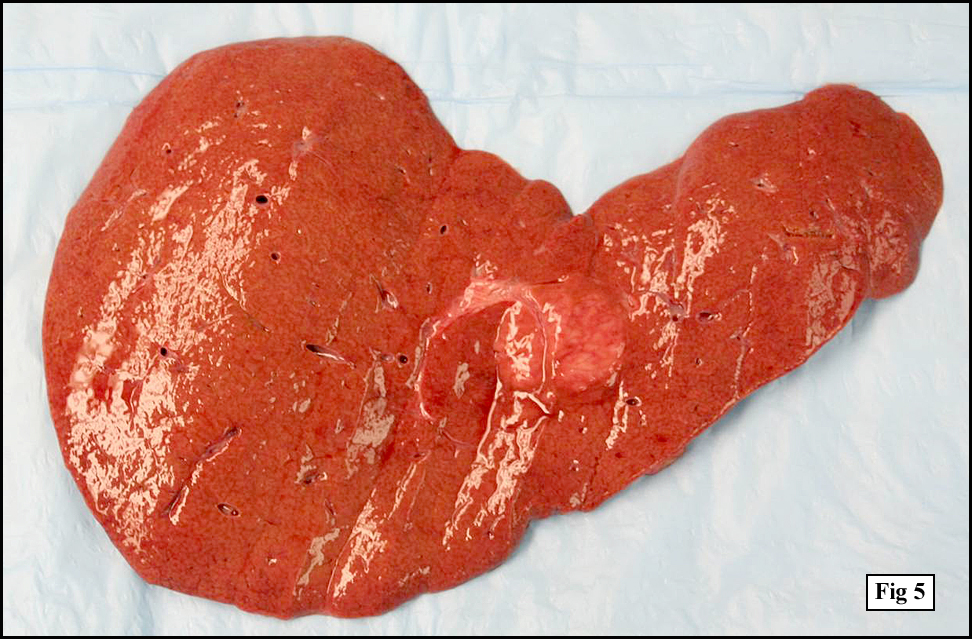
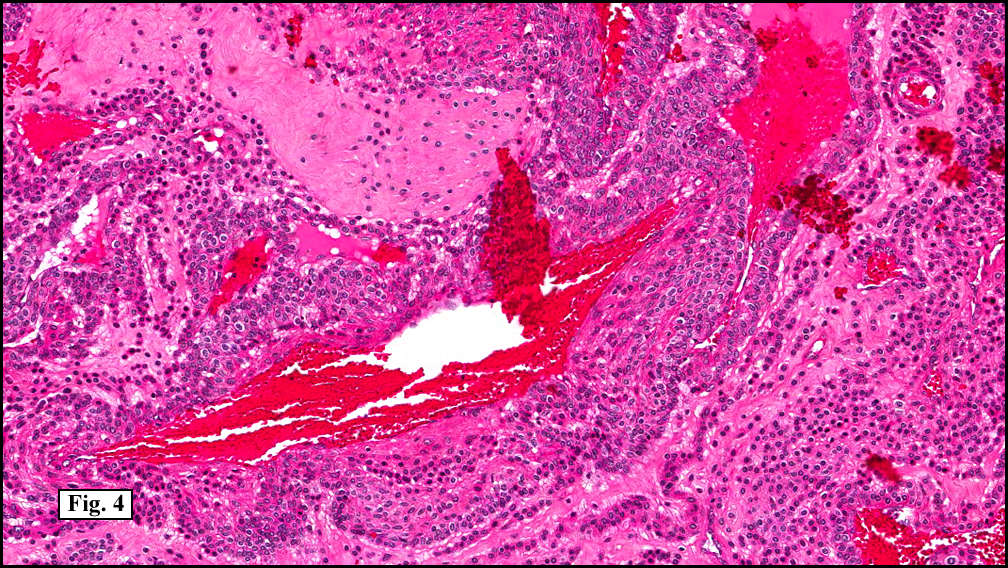
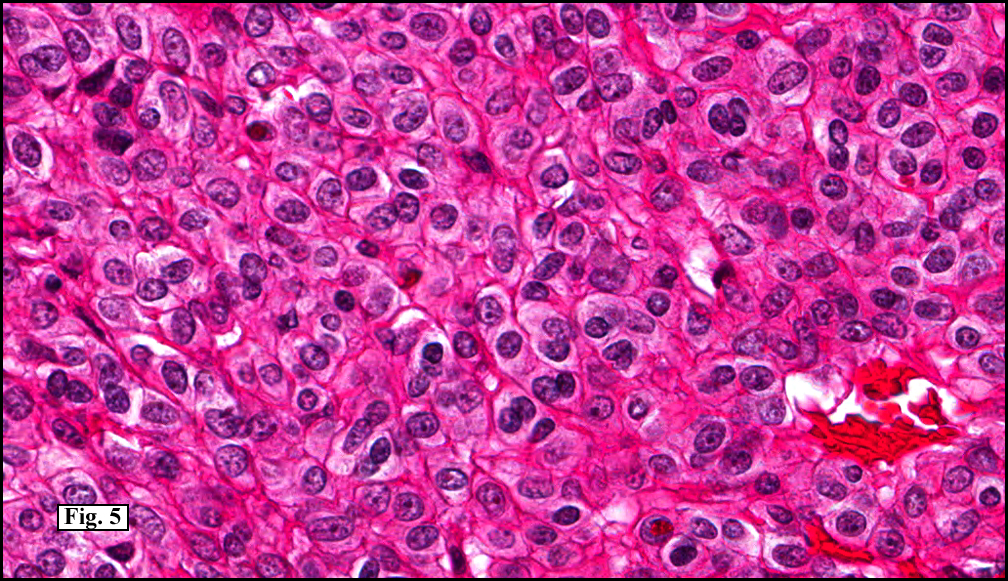
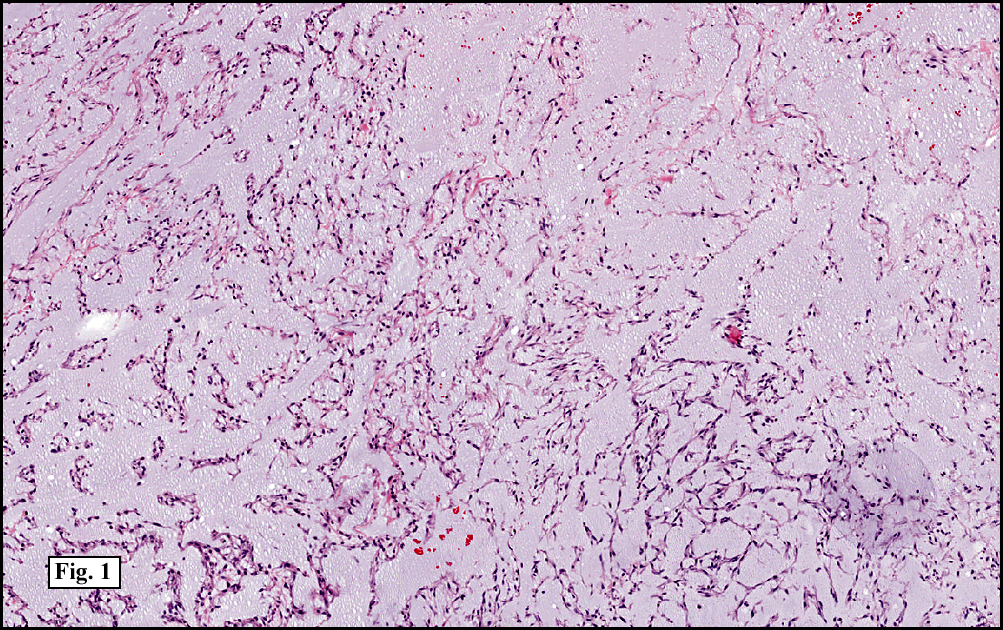
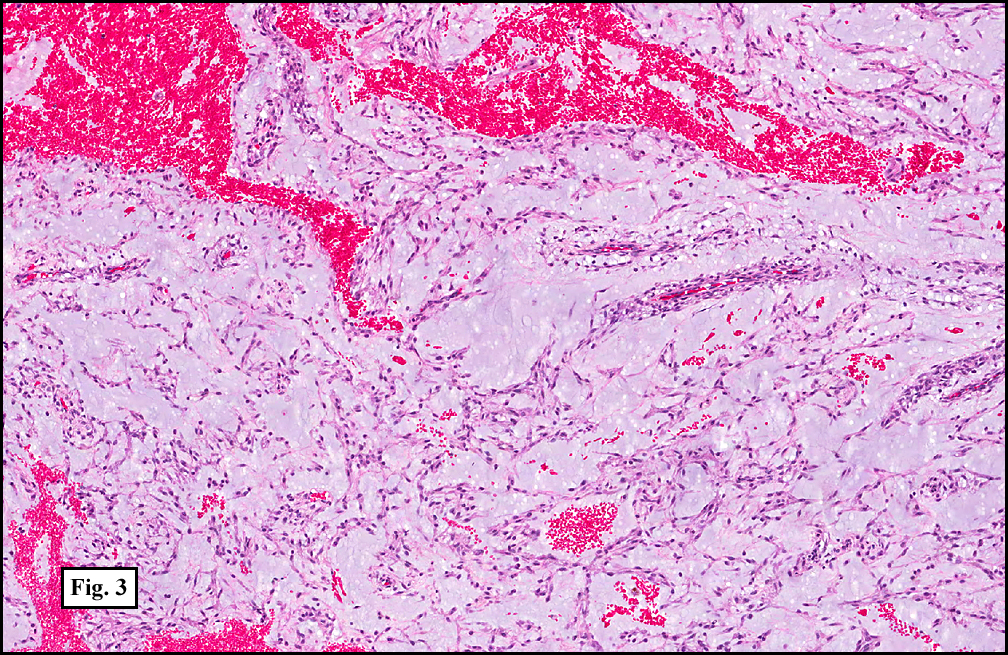
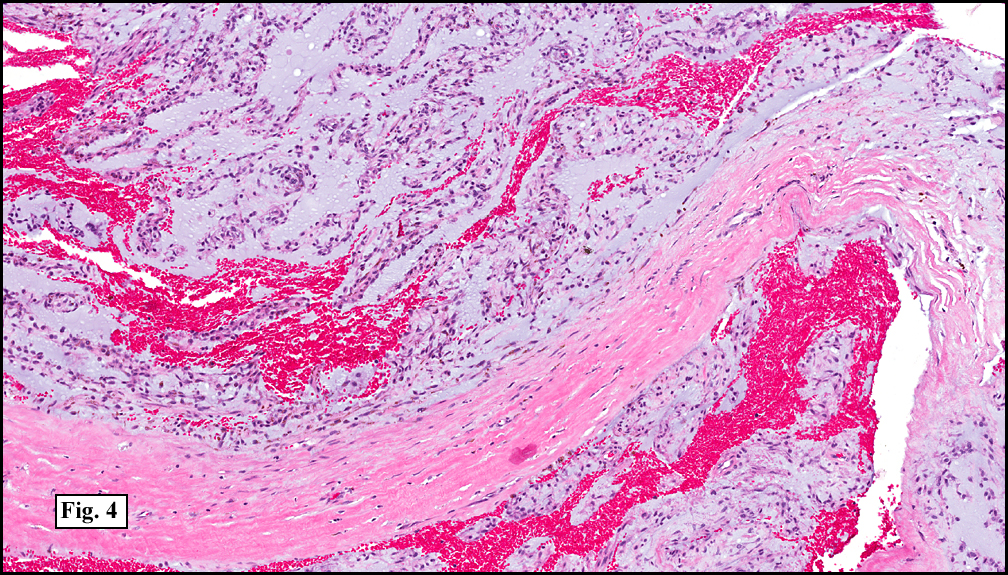
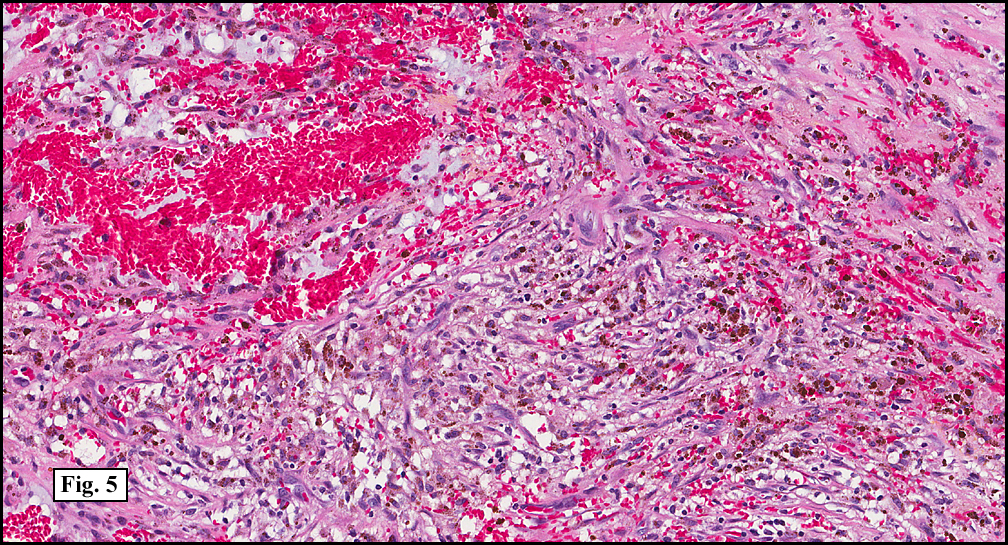
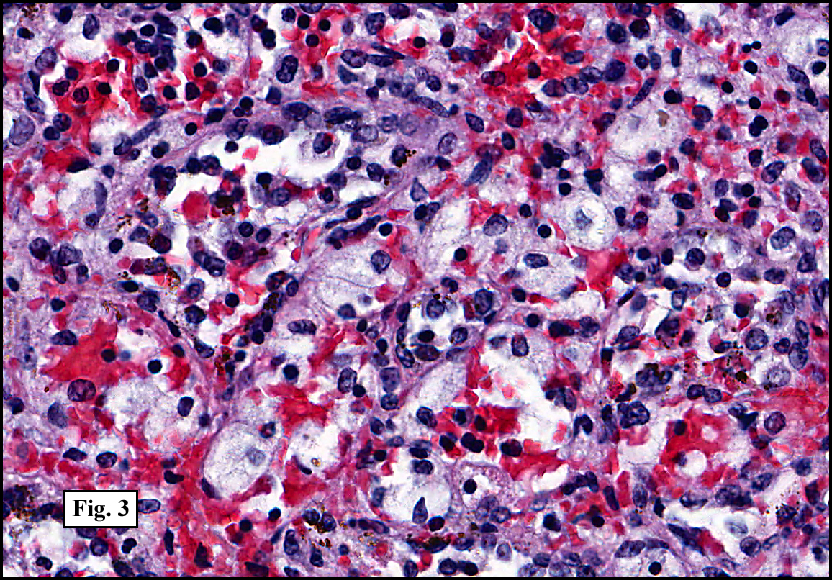
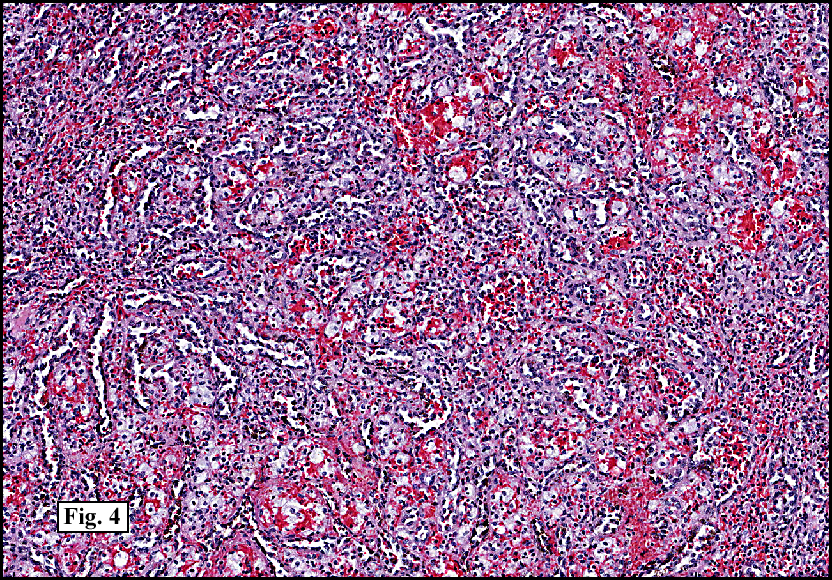
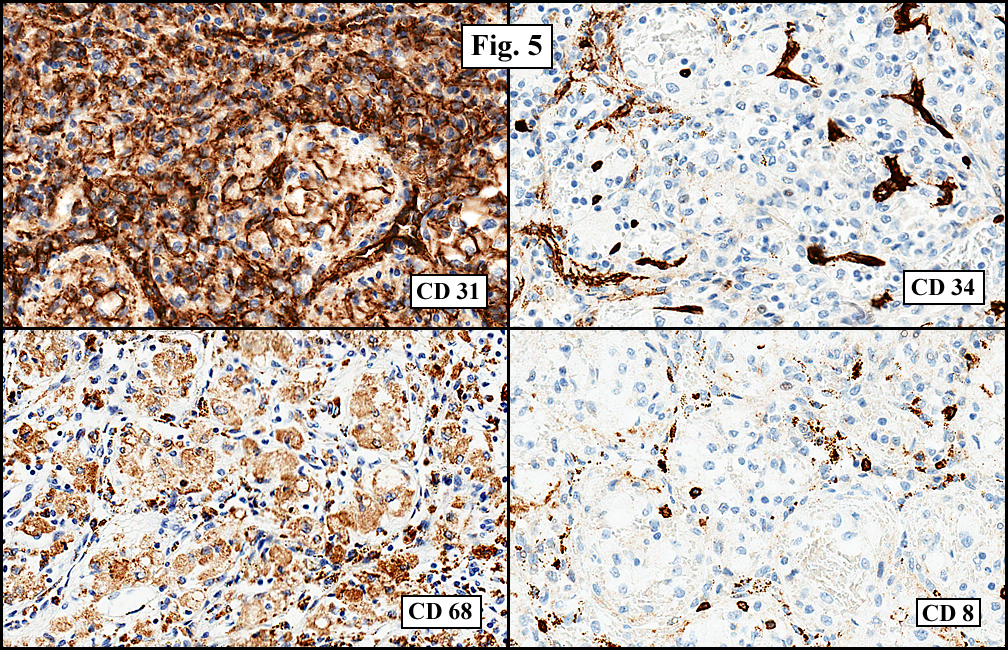
Sections of the gastric mass showed replacement of the lamina propria and distortion of normal gastric glands (Fig. 3) by an infiltrate of monomorphic, medium to large lymphocytes with round nuclei, finely clumped chromatin and multiple basophilic medium-sized, occasionally paracentrally situated nucleoli (Figs. 4, 5). Occasional mitotic figures were seen. Focal superficial erosion with acute inflammation was also present. Giemsa stain was performed and showed no evidence of Helicobacter pylori microorganism. Immunohistochemically, the tumor cells showed expression of CD45, CD20, CD79a, CD10, BCL-6 with a high Ki-67 proliferation index (>95%), but was negative for CD138 and CDX2 (Fig. 6).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
CT studies revealed diffuse lymphadenopathy throughout his abdomen and pelvis. A large left groin mass was also present on physical examination. Needle core biopsies were performed and revealed a morphology similar to the gastric lesion with diffuse replacement of normal nodal architecture by a monomorphic population of medium to large lymphoid cells. A flow cytometric study revealed the lymphocytes to show expression of CD20, CD19, CD10 with lambda restriction. A portion of the biopsy material was submitted for FISH analysis showing a subpopulation of cells with a c-myc gene rearrangement.
A follow-up CT of the abdomen and pelvis performed two years following treatment with R-CHOP showed the stomach mass and all the previously enlarged lymph nodes to have disappeared.
Diagnosis: “Burkitt Lymphomaâ€
Hannah H. Wong, MD, Jeffrey D. Cao, MD, Donald R. Chase, MD
Department of Pathology, Loma Linda University and Medical Center,
Loma Linda, California
California Tumor Tissue Registry, Loma Linda, California
Discussion: Burkitt lymphoma (BL) is a B-cell non-Hodgkin lymphoma which was first described in 1958 by Denis Burkitt in the lymphoid tissue around the tooth of an African child. It comprises up to half of all childhood non-Hodgkins lymphoma; however, is an uncommon form of non-Hodgkin lymphoma in adults, with a reported yearly incidence of 1200 patients in the United States. Although it is generally described to be more common in children and young adult males, the 2007 National Cancer Institute Surveillance, Epidemiology and End Results (SEER) database showed 59% of all adult Burkitt lymphoma cases in the United States were older than 40 years of age. Clinically, Burkitt lymphoma is divided onto three variants based on their clinical presentation, morphology and biology.
• Endemic BL: Most common in children from equatorial Africa, it generally presents in the jaw with eventual spread to extranodal sites, such as the bone marrow and leptomeniges.
• Sporadic BL: There is no geographical limitation to this variant, which is mostly found in children and young adults. The median age in adult patients is 30 years, and there is a slight predilection for males. Sporadic BL most often present as abdominal masses, especially in the ileo-cecal region. However, it may be found in the ovaries, kidneys and breasts. Involvement of lymph nodes has been reported to be more common in adults compared to children.
• Immunodeficiency-associated BL: This variant of BL is primarily limited to patients with human immunodeficiency virus (HIV) infection.
Adult patients with Burkitt lymphoma in the United States most often present with constitutional B symptoms (fever, fatigue, weight loss), bulky abdominal masses and rarely lymph node involvement. The tumor is so aggressive that bone marrow involvement is present at the time of diagnosis 70% of the time, and leptomeningeal involvement presents in up to 40% of patients. A timely diagnosis is vital for this aggressive lesion, and initial therapy is aimed to begin within 48 hours of diagnosis.
Microscopically, there is generalized effacement of normal architecture by a diffuse monotonous population of cohesive, intermediate-sized lymphoid cells with high nuclear to cytoplasmic ratios and smooth round to oval nuclear contours. The nuclei show finely clumped, dispersed chromatin and generally have multiple nucleoli. Frequent mitotic figures and apoptotic bodies are generally present, with frequent benign macrophages filled with ingested apoptotic tumor cells creating the characteristic “starry-sky†appearance. A florid granulomatous reaction may be present in some cases of BL, making recognition of the underlying BL difficult. However, these lesions generally have a good prognosis. The malignant lymphoid cells may also have eccentric basophilic cytoplasm with a single central nucleolus.
Immunohistochemically, the tumor cells generally express B-cell antigens such as CD19, and CD20 as well as CD10, BCL6, CD38, CD77 and CD43. They generally do not express TdT, but may be weakly positive for BCL2. There is generally up to 100% positivity of Ki-67 in most of these lesions. The immunophenotype of BL may be demonstrated by tissue immunohistochemistry or flow cytometry. Gene expression analysis is important in BL, as a large number cases have the diagnostic MYC translocation at band 8q24 of the IG heavy chain region 14q23, t(8;14). Other common translocation patterns in BL include t(2;8) and t(8:22). Despite the aggressive nature of BL, it is a highly curable condition. Patients with BL generally respond well to high doses of systemic chemotherapy, and intrathecal therapy.
The pathogensis of BL has been associated with the Epstein-Barr virus. Virtually all tumor cells in endemic BL show the presence of EBV within their genome, while EBV positivity is present in approximately 30% of sporadic BL, and 25-40% of immunodeficiency-associated BL. However, there is no known association of BL with Helicobacter pylori (H. pylori). In a review of the current literature there were a total of 4 cases of BL associated with H. pylori, with three of those cases being in children and one in a 53-year-old male. H. pylori is a gram negative rod first identified by Warren and Marshall in 1983. H. pylori has been associated with chronic active gastritis, peptic ulcers and subsequent development of carcinoma, and gastric lymphoid neoplasms, specifically mucosa-associated lymphoid tissue (MALT) lymphomas of the stomach. In fact, H. pylori is listed as a Grade I carcinogen by the World Health Organization.
Gastric MALT lymphoma is a marginal zone B-cell low grade lymphoma generally affecting older adults. It is the most common type of extranodal marginal zone lymphoma and comprises up to 85% of extranodal MALT lymphomas. The association between gastric MALT lymphoma and H. pylori has been well established. The stomach in its normal physiologic state does not possess any organized lymphoid tissue. When H. pylori infects the stomach, it produces a host of antigens which stimulate H. pylori-specific T cells to induce proliferation of B lymphocytes. The proliferation of the B cells then leads to the development of lymphoid proliferations of the MALT type. The role of H. pylori in gastric MALT lymphoma has been further established as the lymphoma rapidly regresses with eradication of H. pylori. It has been proposed that the association between gastric BL and H. pylori may be similar to that of H. pylori and gastric MALT lymphoma.
Despite the possibility of a similar causal relationship between H. pylori and Burkitts lymphoma, BL and gastric MALT lymphoma are quite different entities. Patients with gastric MALT lymphoma often present with abdominal pain, and stage I or II disease unlike BL which is generally stage IV at the time of diagnosis. Bone marrow involvement is rare unlike BL. Microscopically, the lymphoma cells are small to medium in size with slightly irregular nuclei, moderately dispersed chromatin and inconspicuous nucleoli and generally abundant pale cytoplasm; in contrast to BL where the lymphoma cells have scant cytoplasm and multiple prominent nucleoli. The lymphoma cells of gastric MALT lymphoma express CD20 and CD79a but generally do not express CD5, CD10, CD23 and have variable expression for CD43 and CD5. Gastric MALT lymphomas are associated with various translocations such as t(11;18). Treatment for this entity generally starts with treatment of H. pylori by antibiotics, and this has been found to be successful for many cases; however, chemotherapy may be required in some aggressive cases.
Suggested Reading:
Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele N, Vardiman JW, Eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: IARC; 2008.
Grewal SS, Hunt JP, O’Connor SC, Gianturco LE, Richardson MW, Lehmann LE. Helicobacter pylori associated gastric burkitt lymphoma. Pediatr Blood Cancer. 2007;10:888-890.
Shannon C, Vickers C, Field A, Ward R. Burkitt’s lymphoma associated with Helicobacter pylori. Journal of Gastroenterology and Hepatology. 2000;15:99-103.
Perkins AS and Friedberg JW. Burkitt lymphoma in adults. Hematology. 2008:341-348
Andoh A, Takaya H, Bamba M, Sakumoto H, Inoue T, Tujikawa T, Koyama S, Fujiyama Y, Bamba T. Primary gastric burkitt’s lymphoma presenting with c-myc gene rearrangement. J Gastroenterol. 1998;33:710-715.
Moschovi M, Menegas D, Stefanaki K, Constantinidou CVV, Tzortzatou-Stathopoulou F. Primary gastric burkitt lymphoma in childhood: associated with helicobacter pylori? Med Pediatr Oncol. 2003;41:444-447.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}