History: A 77-year-old man was found to have a 3.5 x 3.0 x 2.0 cm anterior mediastinal mass. At surgery it was smooth and well-circumscribed. The cut surface was pale tan and lobulated.
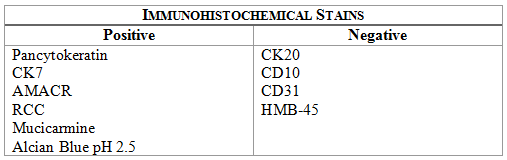
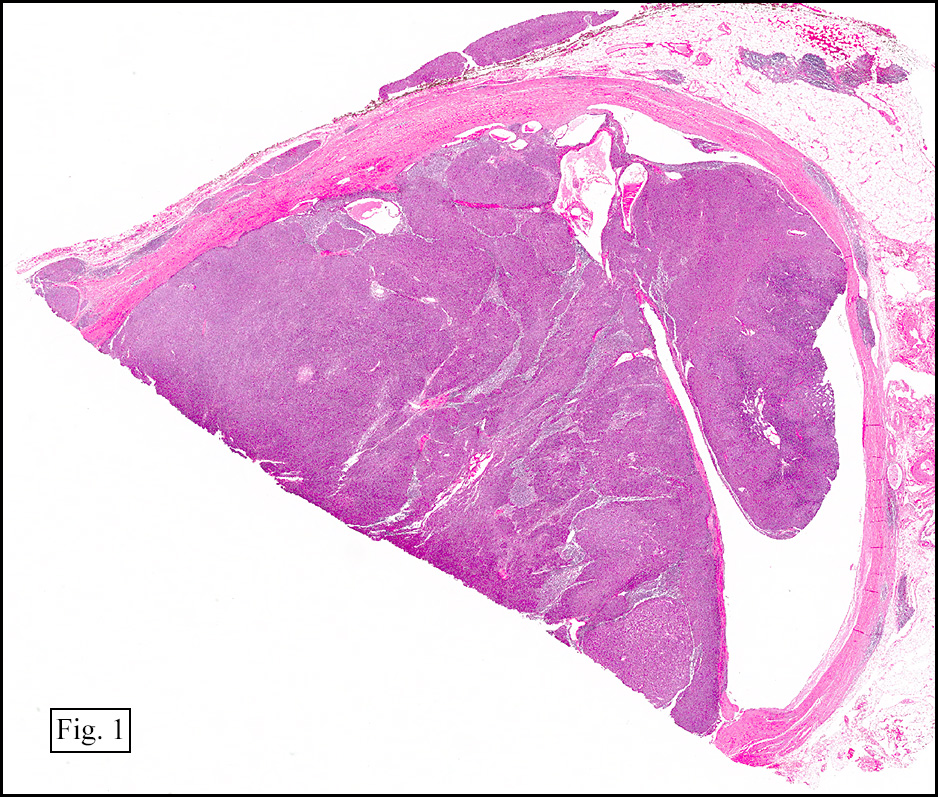
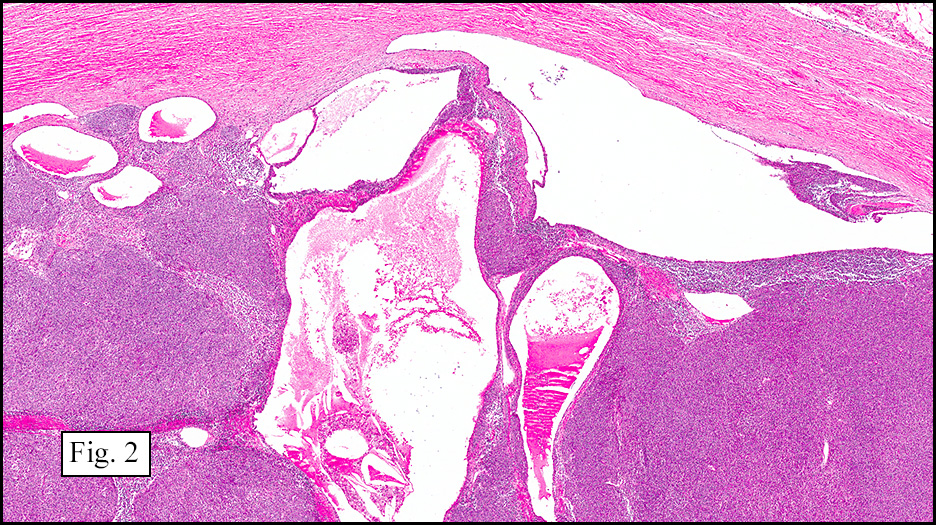
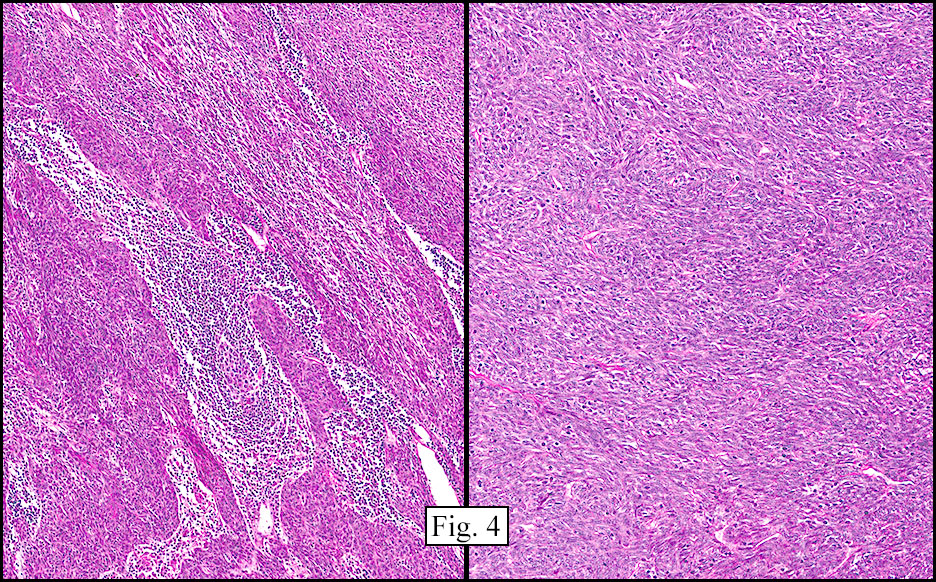
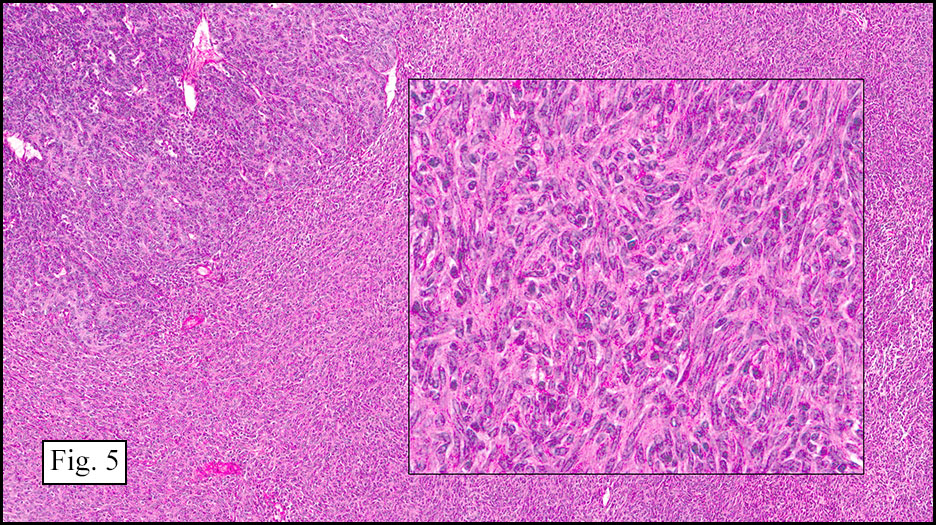
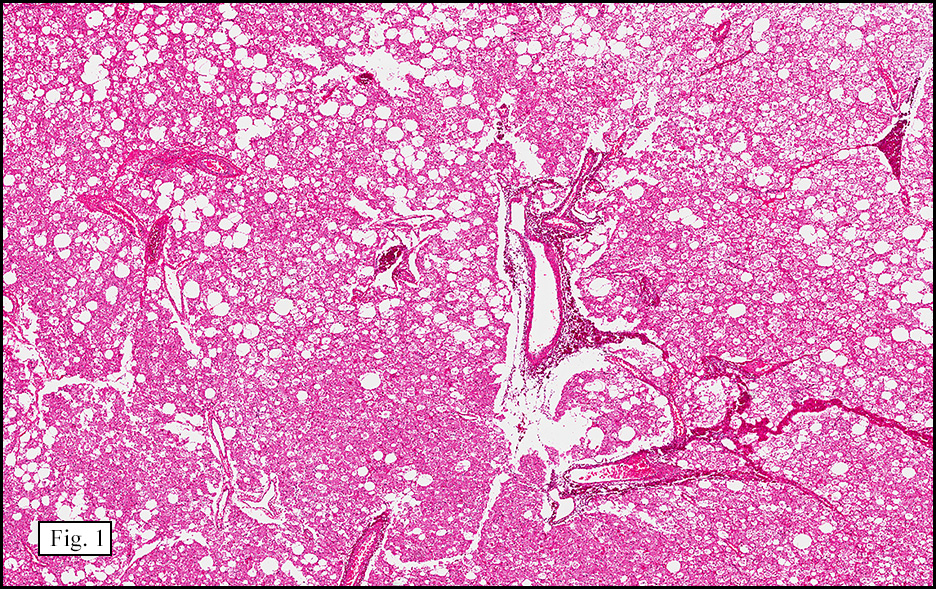
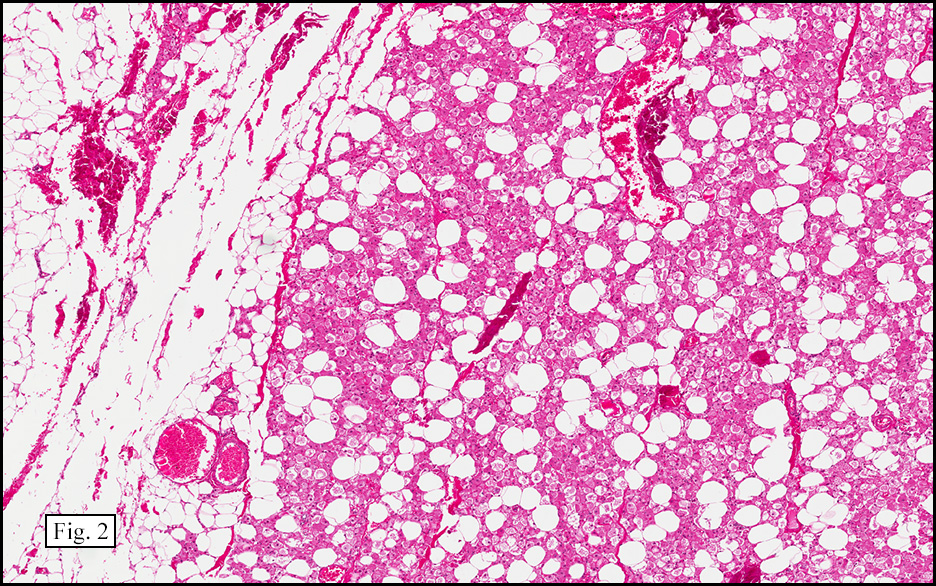
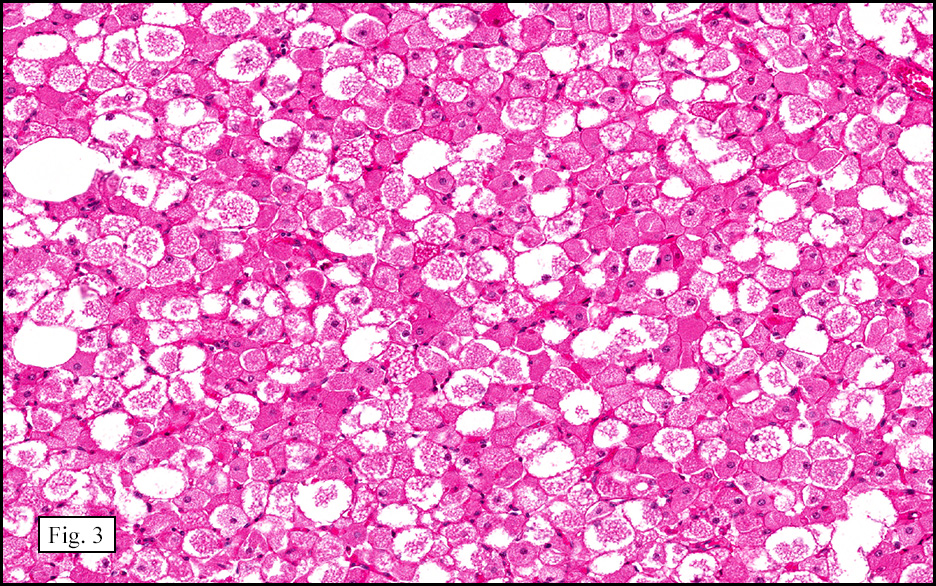
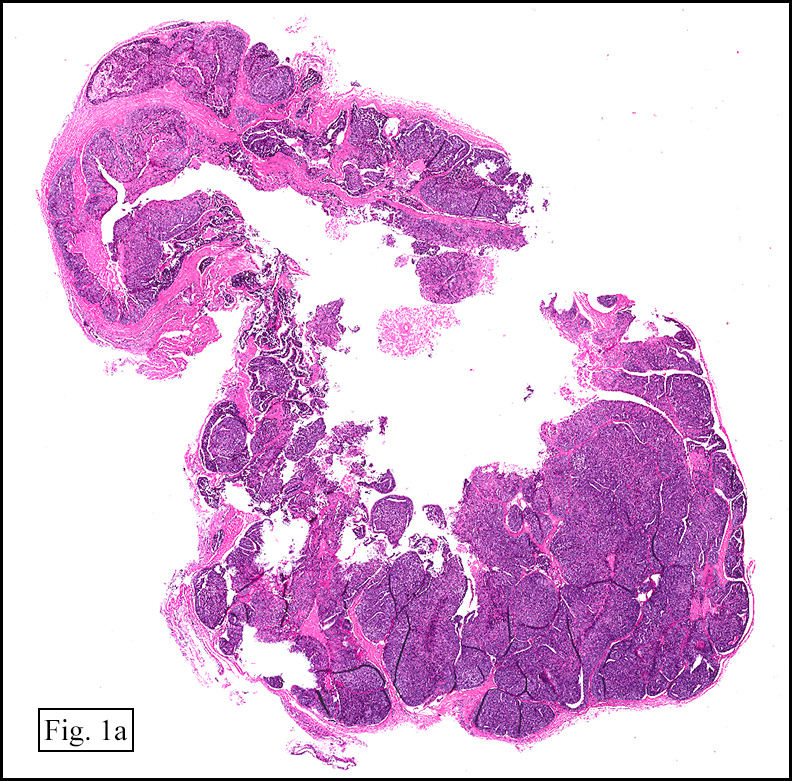
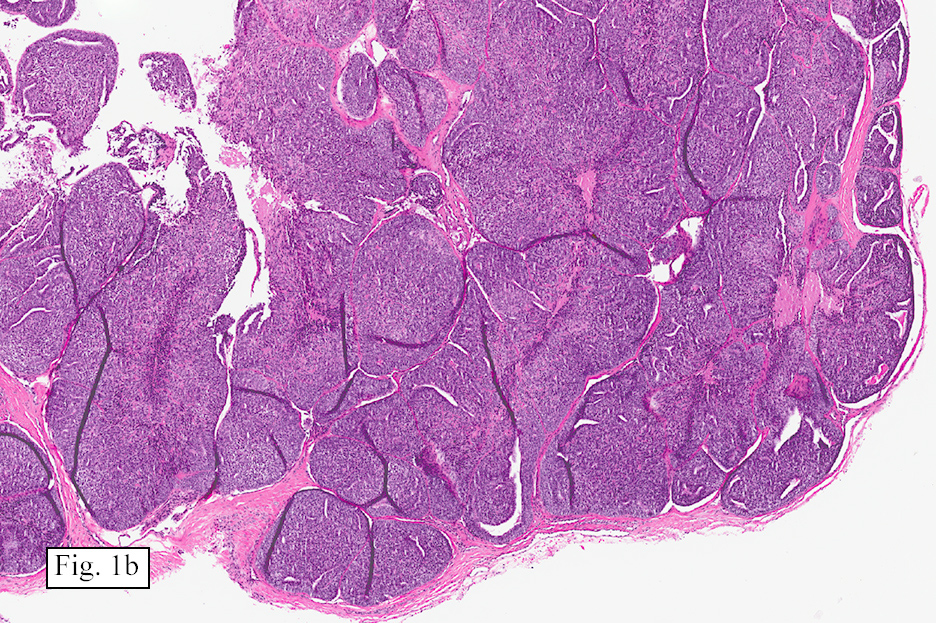
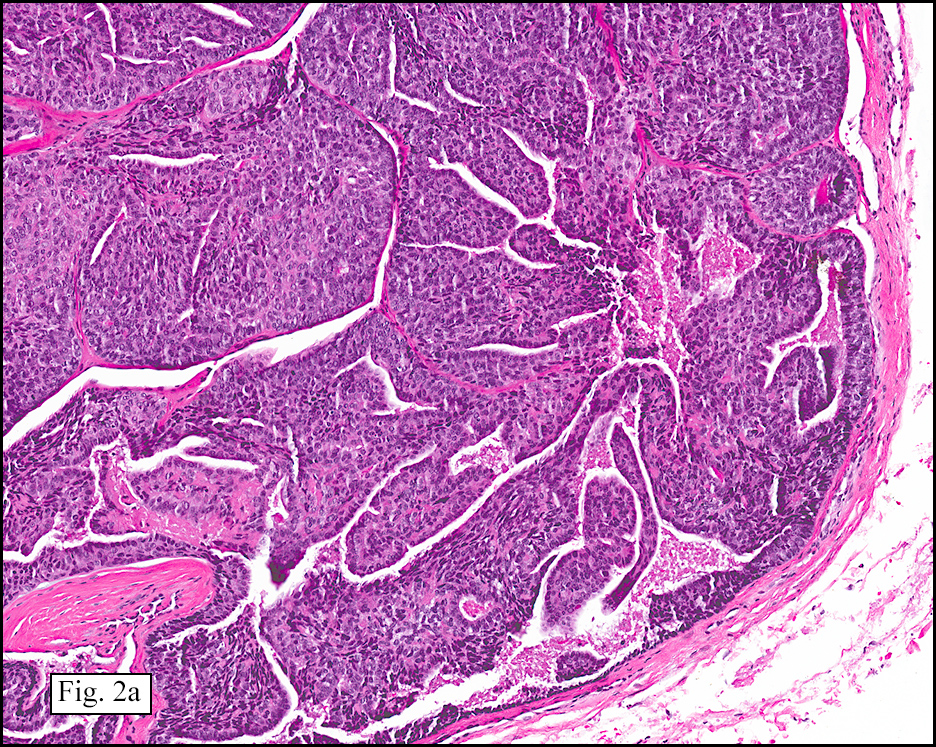
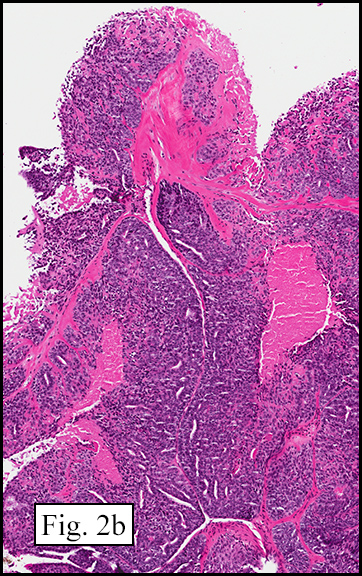
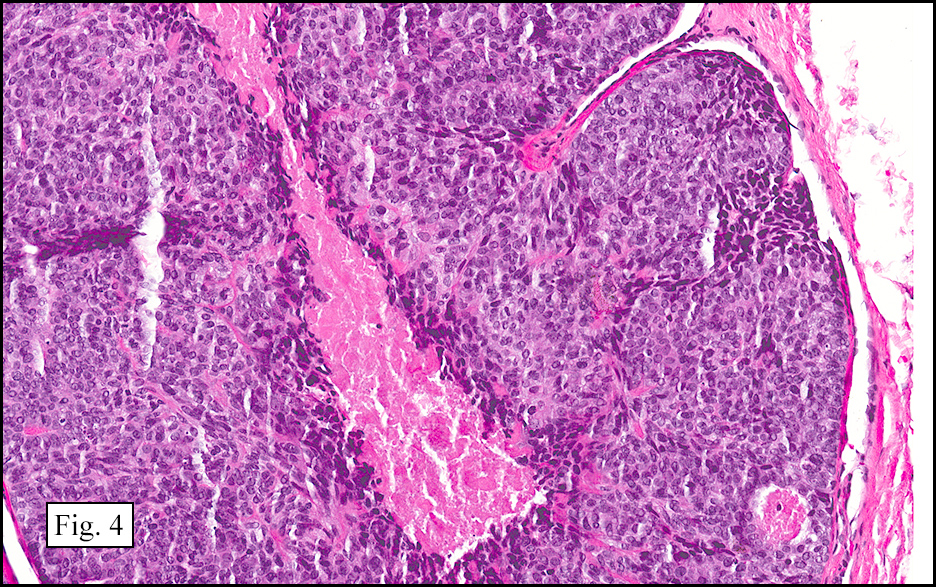
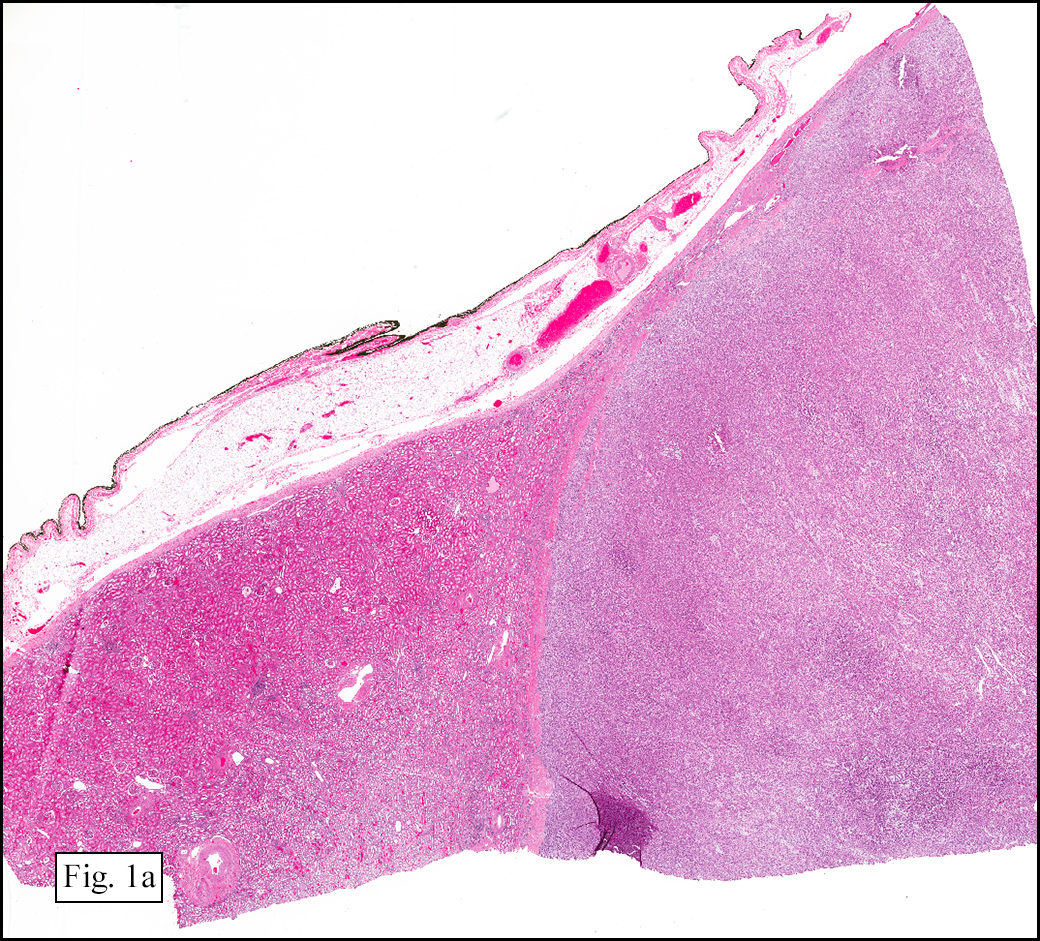
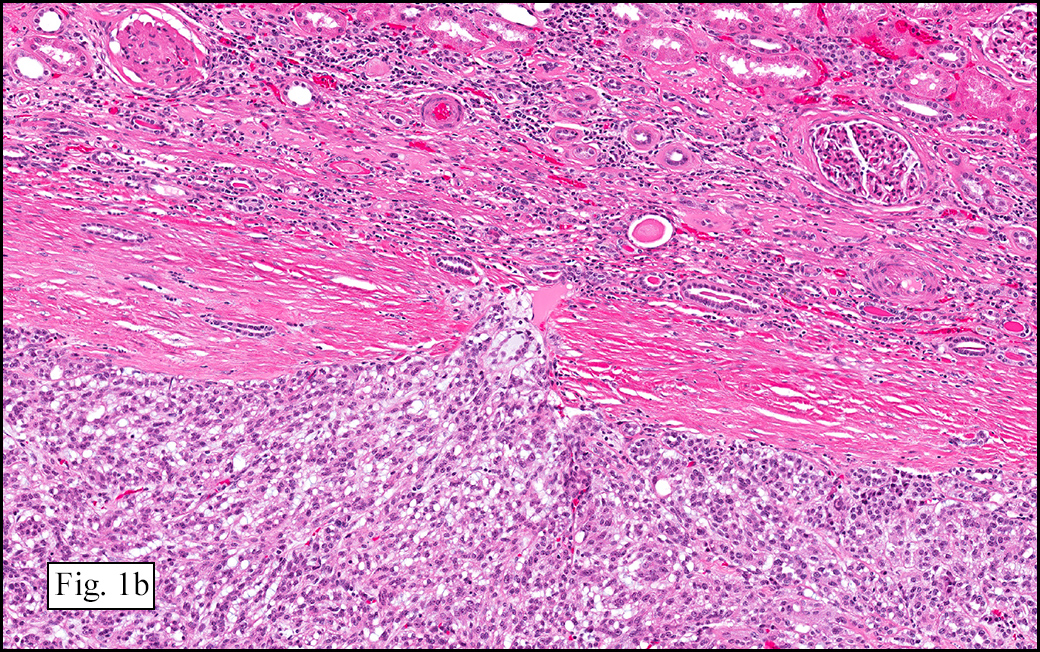
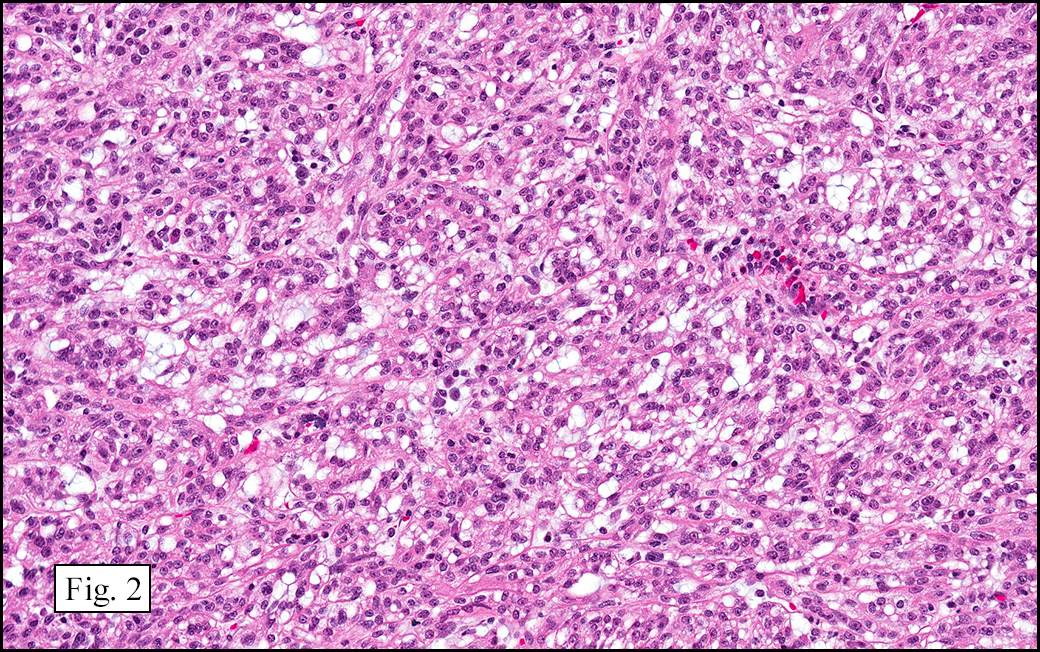
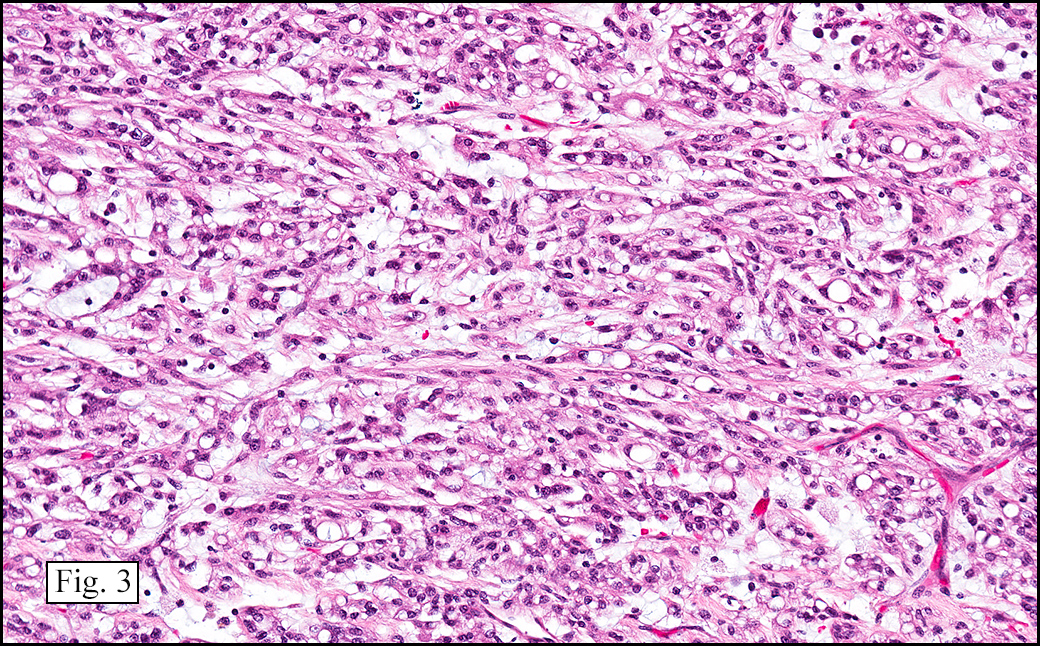
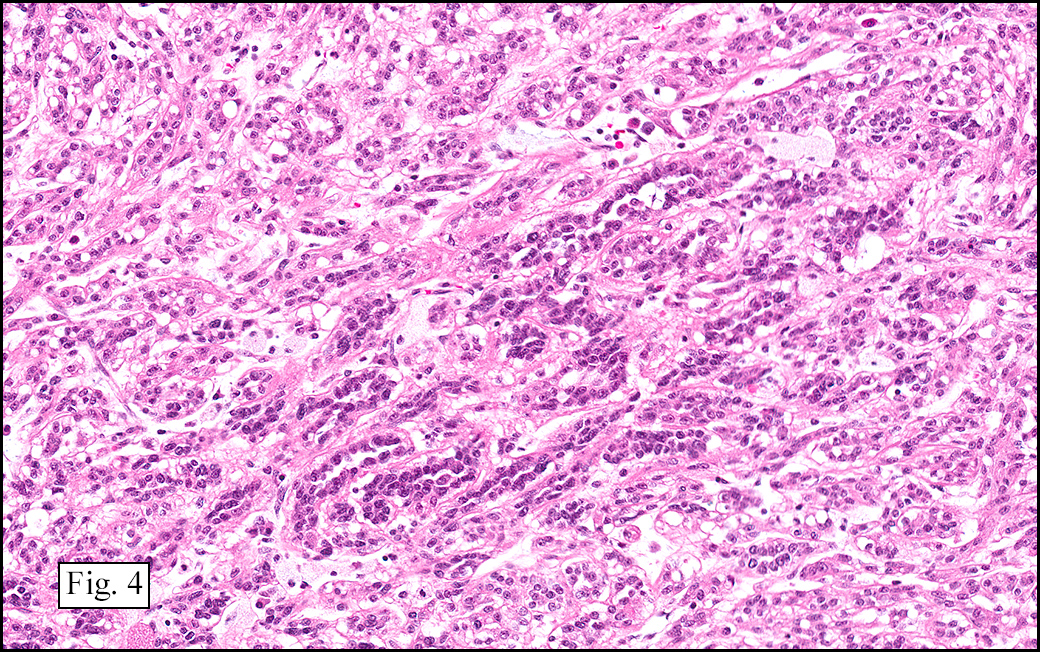
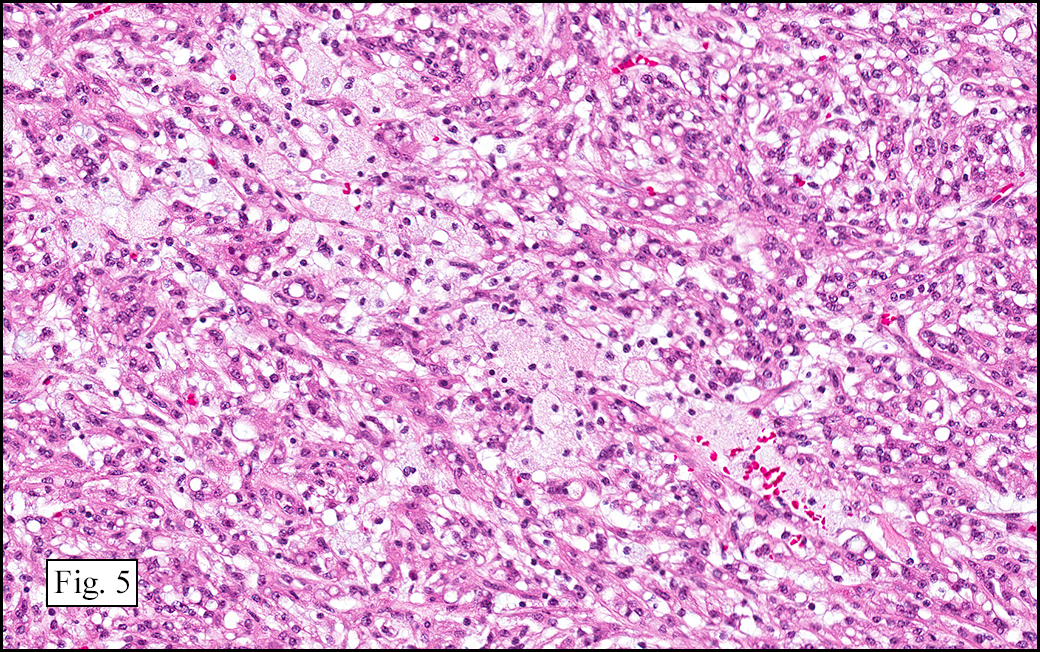
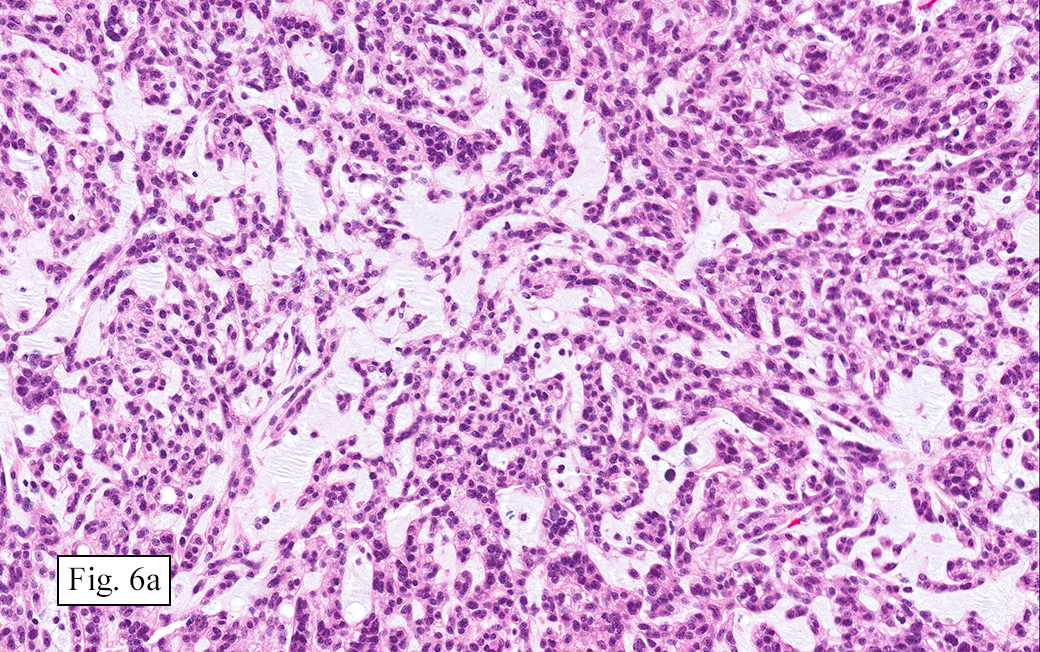
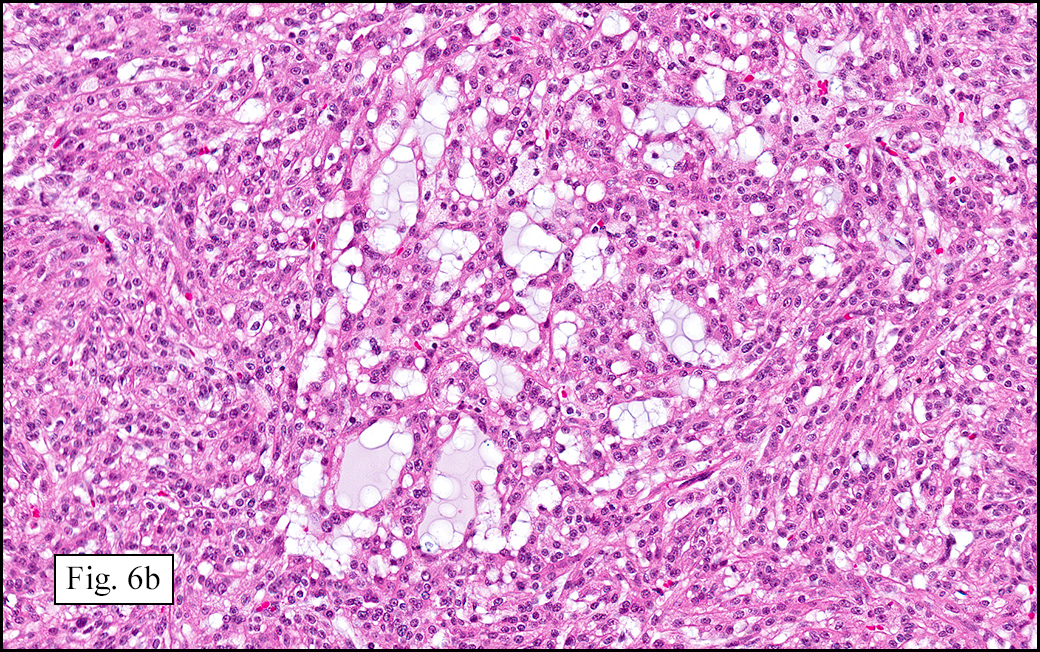
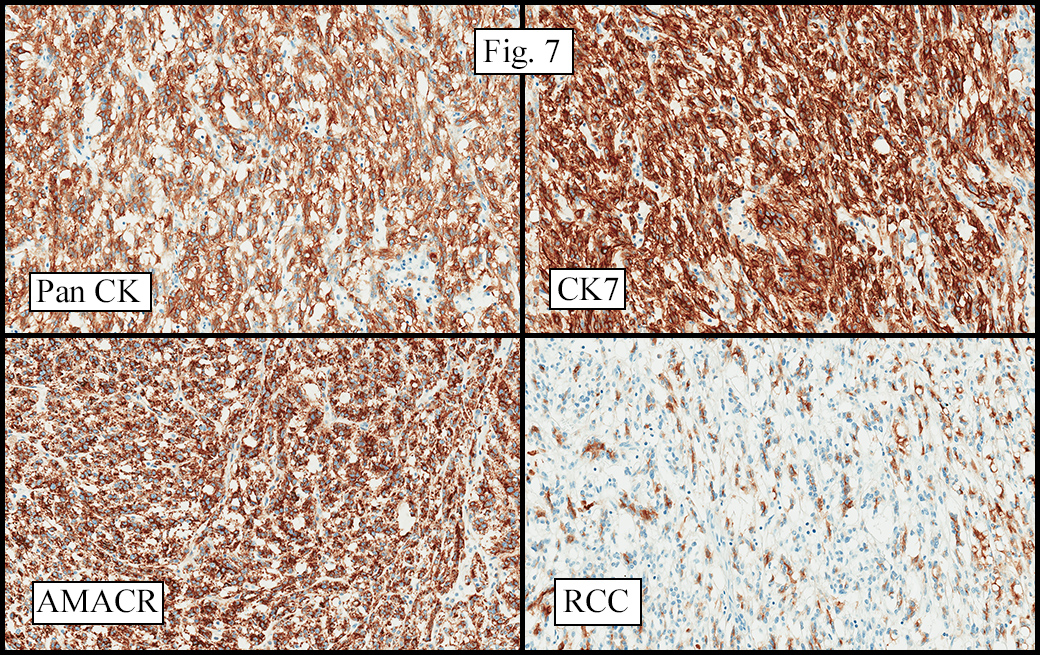
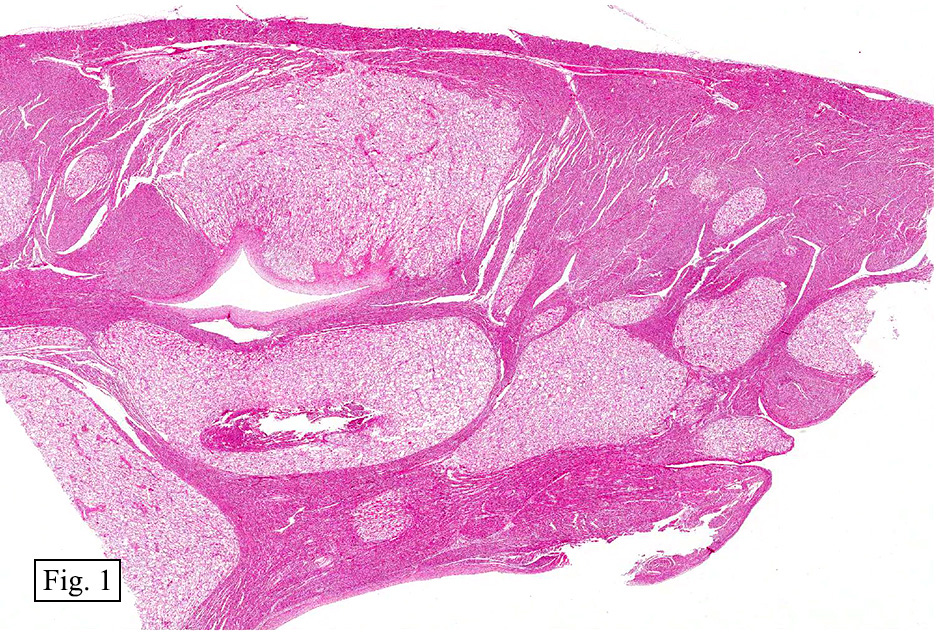
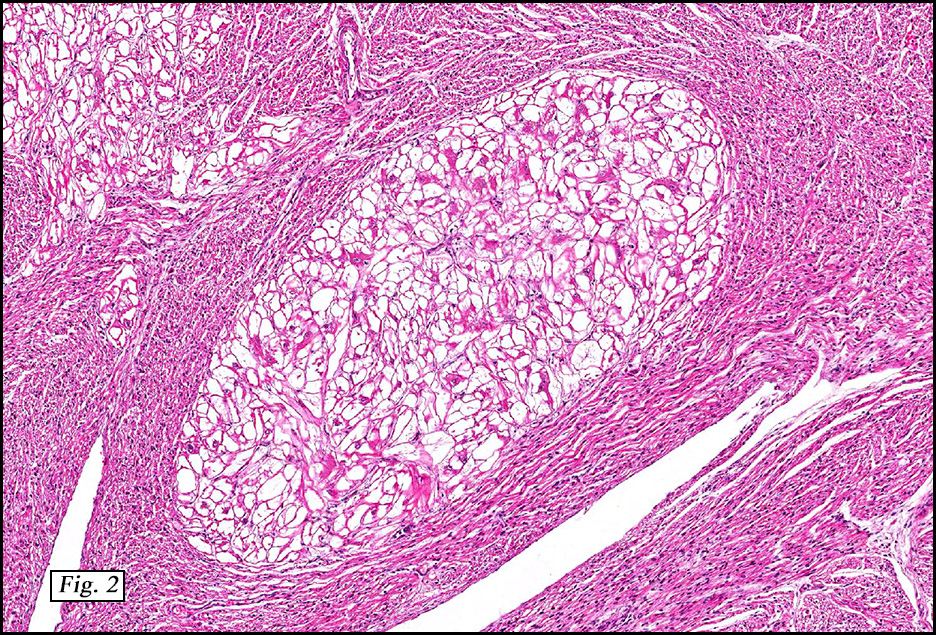
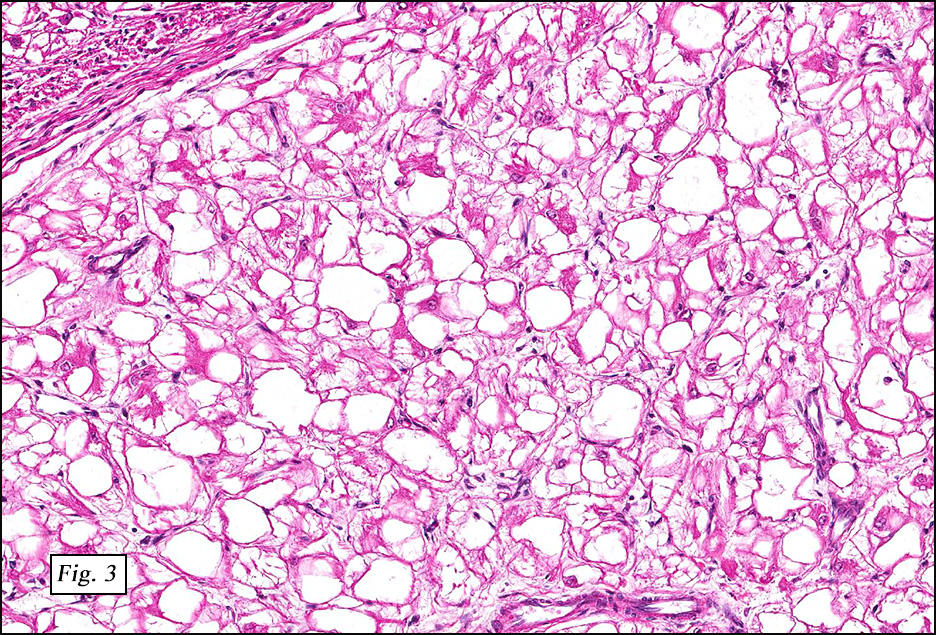
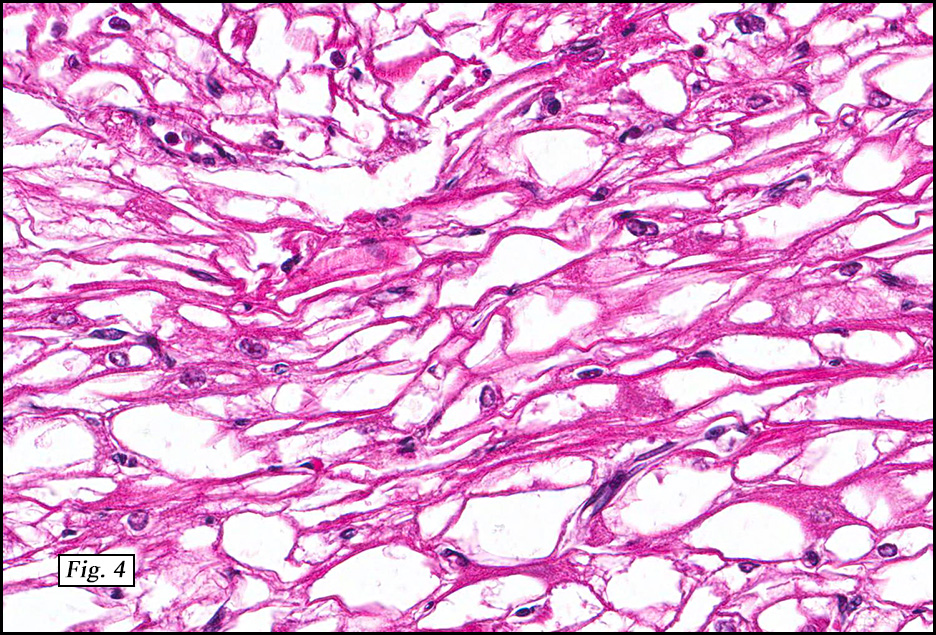
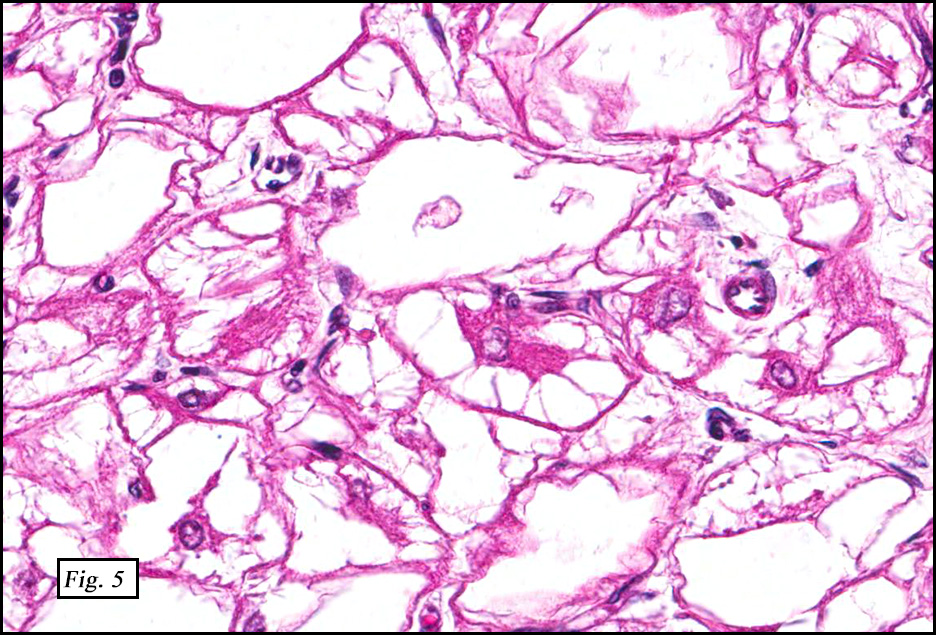
Microscopically, the tumor was encapsulated (Fig. 1) and focally cystic (Fig. 2), mostly consisting of small round to oval lymphocytes (Fig. 3). A prominent spindled component was also present (Fig. 4. Generally the cells had scant cytoplasm and stippled nuclear chromatin. Nucleoli were absent or inconspicuous (Fig. 5). By immunohistochemistry, most of the cells marked as immature T-lymphocytes.
Diagnosis: Spindle cell thymoma (WHO type A)
Summer Blount MD, Donald R. Chase MD
Department of Pathology and Human Anatomy,
Loma Linda University Medical Center, Loma Linda, California
California Tumor Tissue Registry, Loma Linda, California
Discussion: “Thymoma†generically describes a tumor of the thymus, irrespective of the cell of origin or degree of malignancy. Throughout the years, knowledge of thymic epithelial cells and lymphocytes has advanced, resulting in several classification schemes. Today thymoma is defined as a benign or low-grade malignant tumor of thymic epithelium with characteristic histologic features, frequently associated with a variable population of immature, but non-neoplastic lymphocytes. So-called “thymic carcinoma†is separated out as a different entity due to differences in both morphology and behavior.
Thymoma is the most common neoplasm of the anterior mediastinum. It occurs in all ethnic groups, ages and sex with no particular predilection. Some thymomas occur in children, but they are more frequently seen in adults, particularly during the fifth and sixth decades of life. The majority is found in the normal thymic location, but due to variations in embryonic descent, the tumor locations may range from the submandibular region to the diaphragm. Symptoms of a thymoma are usually vague but may include dysphagia, hoarseness, chest pain, superior vena cava syndrome, or pleural effusion. There is a fairly common association with myasthenia gravis, as well as a less common association with pure red cell hypoplasia and hypogammaglobulinemia.
Microscopically thymomas are usually at least partially encapsulated by dense fibrous connective tissue. The lobules of thymoma vary widely in size and shape and generally have distinct, but irregular boarders. The formation of perivascular spaces is a characteristic feature seen in approximately two thirds of thymomas. These spaces may contain mature T- and B-lymphocytes, plasma cells and/or mast cells; the center vessel often harbors foamy macrophages and has a tendency to hyalinize.
Imaging, including plain radiographs and computerized tomography (CT), may be helpful in the diagnosis of thymomas. The tumor may be homogeneous or have cystic changes with or without calcifications. CT scans are useful in assessing involvement of surrounding structures. These findings, however, are not specific for thymoma, and may be seen in a variety of other mediastinal tumors including: germ cell tumors, carcinoid tumor and malignant lymphoma.
Thymomas have a morphologic spectrum ranging from tumors consisting almost entirely of lymphocytes to tumors having diffuse growth of atypical polygonal epithelial cells or short spindled epithelial cells. The WHO summarizes this spectrum as follows:

Type A, also known as “spindle cell†or “medullary thymoma†is composed of neoplastic thymic cells having a spindled to ovoid shape without nuclear atypia. These cells may be densely packed in fibroblast-like bundles having a storiform pattern or are loosely organized in ill-defined arrangements. Few or no lymphocytes are present in the background. This subset of thymoma can form a variety of histologic structures including a hemangiopericytoma-like pattern due to dilated sinusoidal vasculature, rosettes, or small glandular spaces that harbors eosinophilic material in the lumen. Additionally the Type A thymoma may have scattered cystic to pseudoglandular spaces with rare glomeruloid structures.
Type AB or “mixed thymoma†is composed of a variable mixture of lymphocyte-poor and lymphocyte-rich thymoma types. In addition to lymphocytes there are small round to spindled epithelial cells with dispersed chromatin and inconspicuous nucleoli. There are no defined criteria as to how much of the two components need to be present in order to diagnosis Type AB thymoma.
Type B1 or “lymphocyte-rich thymomaâ€, sometimes also referred to as “predominately cortical†or “organoidâ€, resembles the normal functional thymus. It is composed of sheets of mature lymphocytes resembling thymic cortex with epithelial cells scattered in areas of immature lymphocytes and areas of medullary differentiation. Hassall’s corpuscles may or may not be present.
Type B2 or “cortical thymoma†is composed mostly of large polygonal cells with vesicular nuclei and prominent nucleoli that resemble normal thymic epithelium. The tumor cells are arranged loosely with numerous intermixed immature T-lymphocytes. Commonly, perivascular arrangement of the tumor cells results in a palisaded appearance. This tumor is sometimes also referred to as a “mixed lymphocytic and epithelial thymoma without medullary differentiationâ€.
Type B3 thymoma may also be referred to as: “epithelialâ€, “atypicalâ€, “squamoidâ€, or “well-differentiated thymic carcinomaâ€. The predominant composition is of medium-sized round to polygonal epithelial cells having mild nuclear atypia. The epithelial cells are mixed with a minor component of intraepithelial lymphocytes, resulting in sheet-like growth.
Not discussed here are rarer subtypes of thymoma referred to as “metaplasticâ€, “microscopicâ€, “sclerosingâ€, or “lipofibroadenomatous†variants.
Suggested Reading:
Shimosato Y, Mukai K, Matsuno Y. AFIP Tumors of the Mediastinum. Series 4. Vol 11. Maryland: ARP Press, 2010: 19-108
Diagnosis and subclassification of thymoma by minimally invasive fine needle aspiration directed by endobronchial ultrasound: a review and discussion of four cases. Moonim MT, Breen R, Gill-Barman B, Santis, G. Cytopathol, Aug 01, 2012; Vol. 23, No. 4, p. 220-228
Molecular Analysis of Thymoma. Badve S, Goswami C, Gökmen-Polar, Y, Nelson Jr. RP, Henley J, Miller N, Zaheer, Narjis A; Sledge Jr. GW, Lang L, Kesle KA, Loehrer Sr. PJ, van Diest P. PLoS ONE, Aug 01, 2012; Vol. 7, No. 8, p. 1-8
Cytokeratin profiles of the thymus and thymomas: histogenetic correlations and proposal for a histological classification of thymomas. Kuo K. Histopathol, May 01, 2000; Vol. 36, No. 5, p. 403-414
Invasive Spindle Cell Thymomas (WHO Type A). Moran CA, Kalhor N, Suster S. Am J Clin Pathol, Nov 01, 2010; Vol. 134, No. 5, p. 793-798
Thymoma: Current Concepts. Kalhor N, Moran CA. Oncology (08909091), Oct 01, 2012; Vol. 26, No. 10, p. 975-981
Tumours and tumour-like conditions of the thymus other than thymoma; a practical approach. den Bakker MA, Oosterhuis, JW. Histopathol, Jan 01, 2009; Vol. 54, No. 1, p. 69-89
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}