History: A 25-year-old woman presented with a mass on her abdominal wall for almost one year. It was excised, and consisted of homogeneous gray-tan soft tissue fragments.
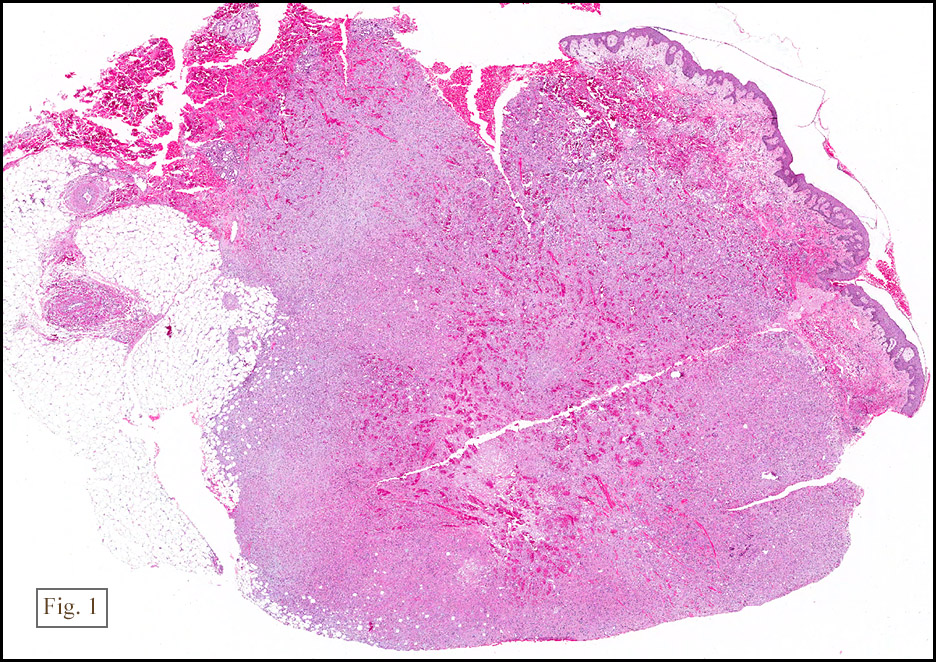
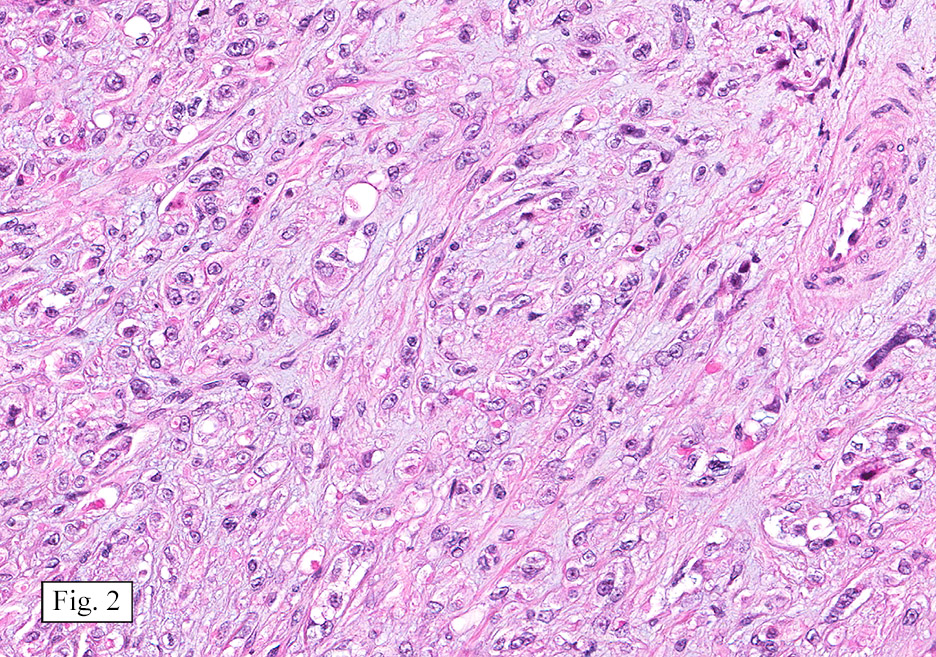
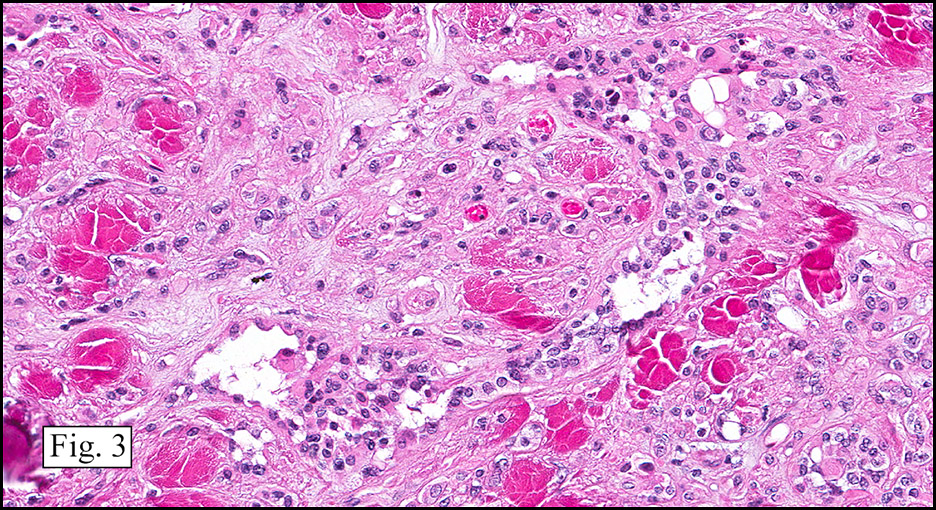
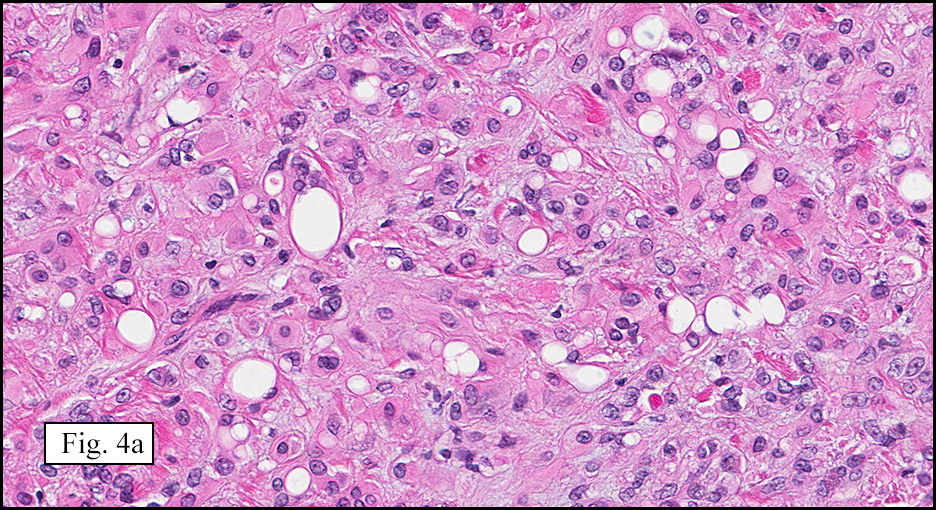
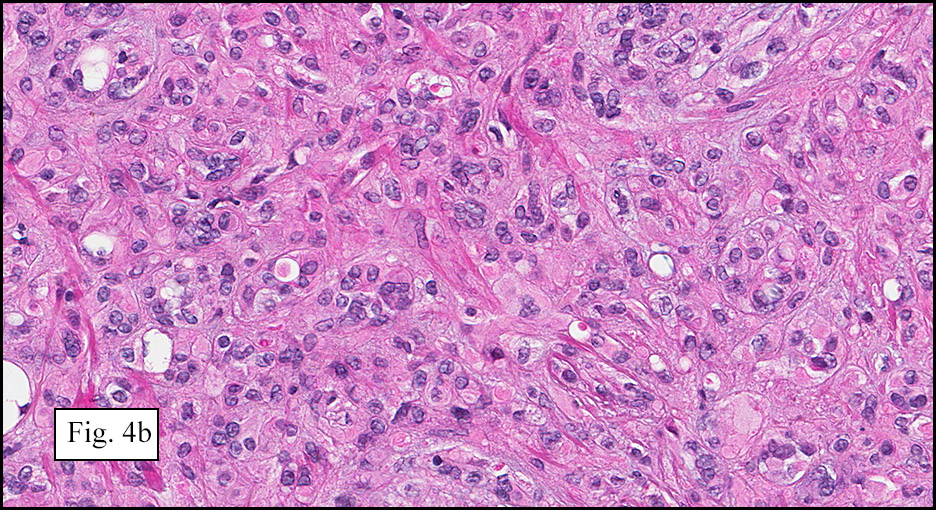
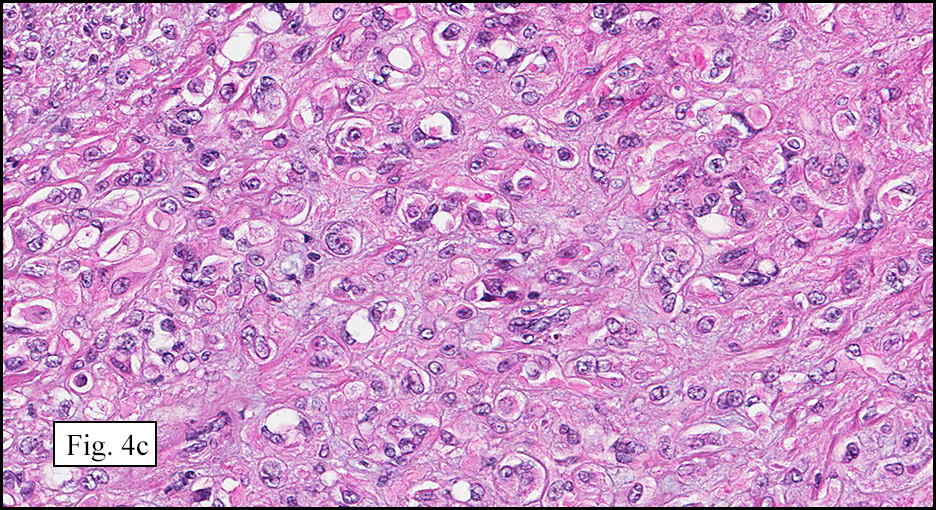
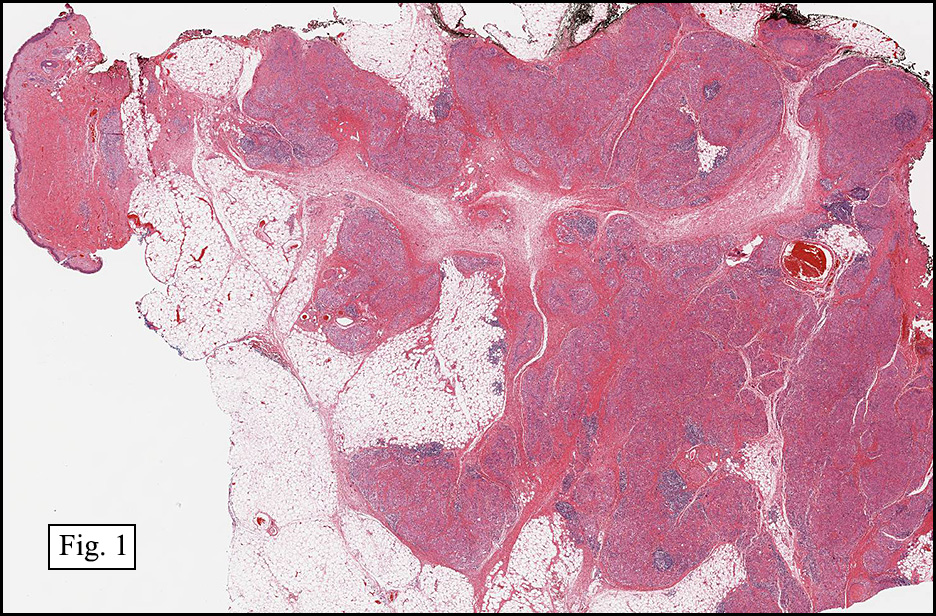
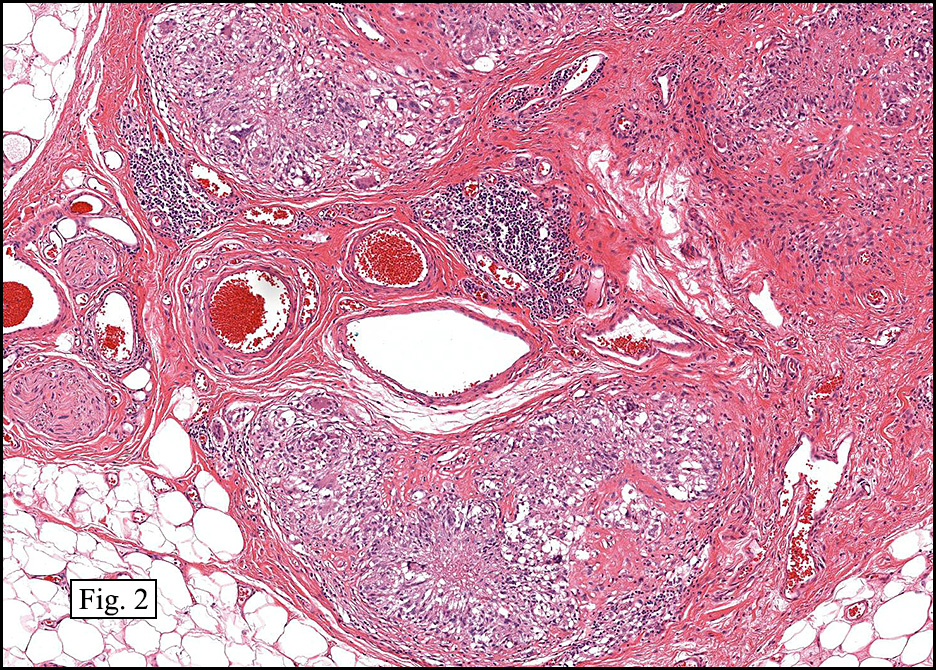
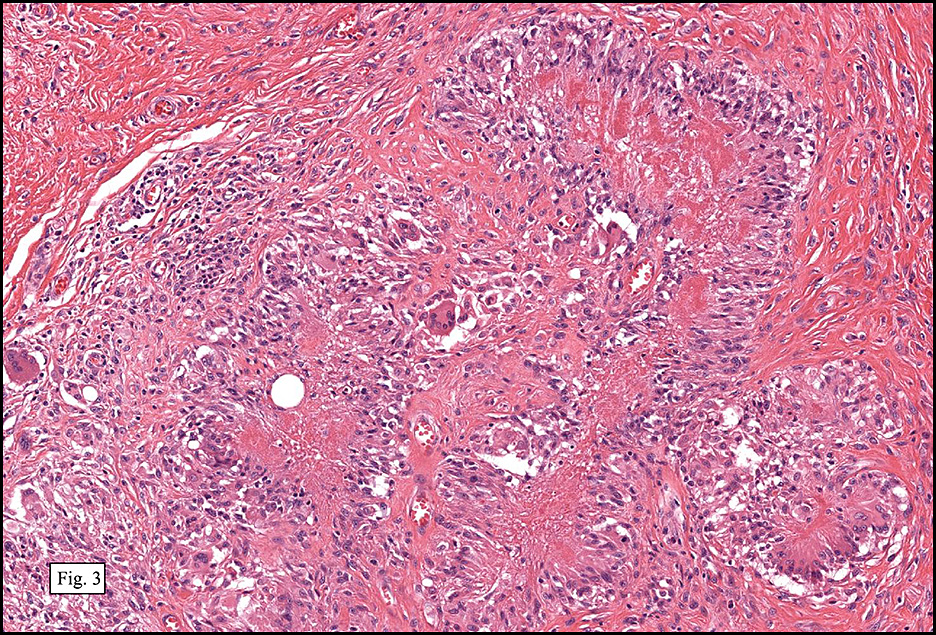
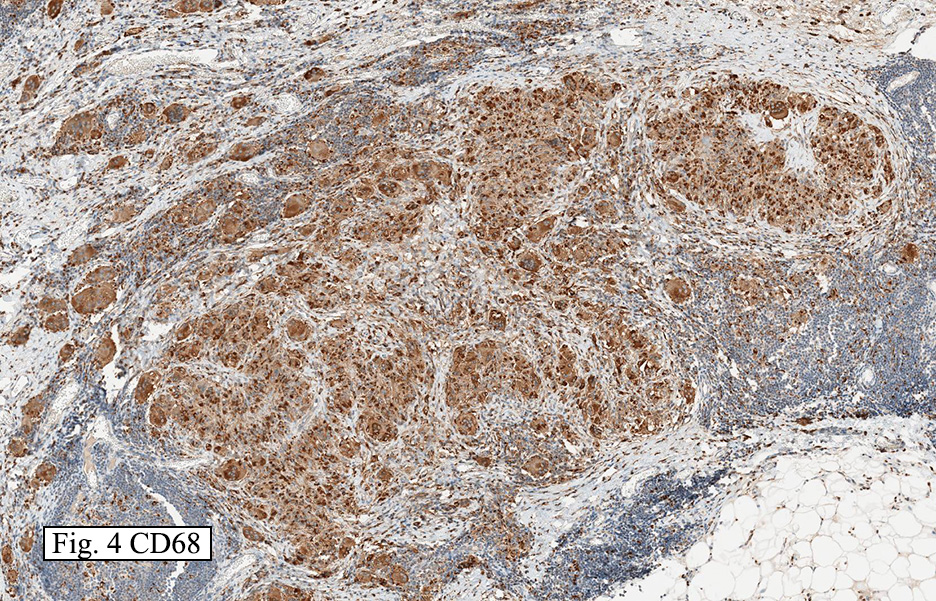
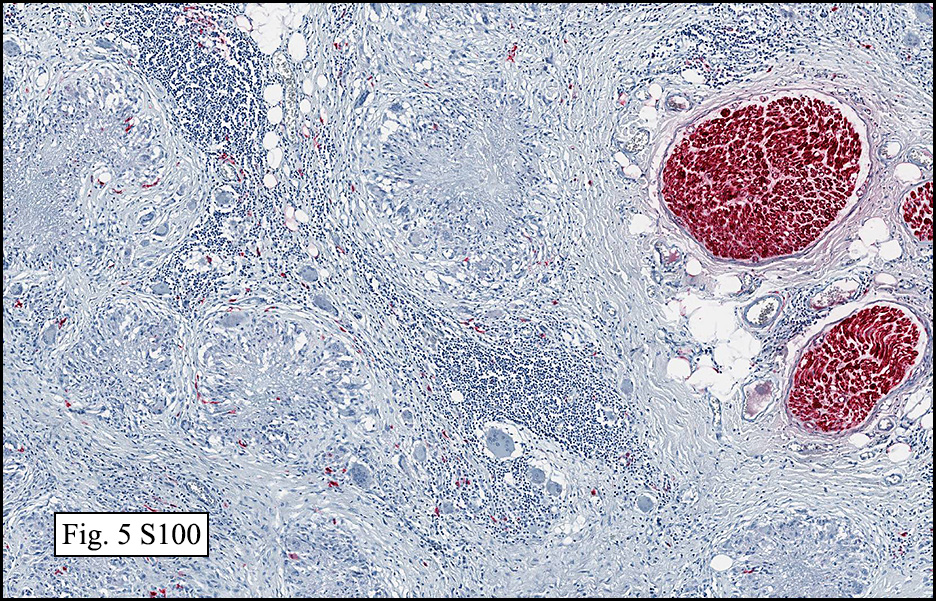
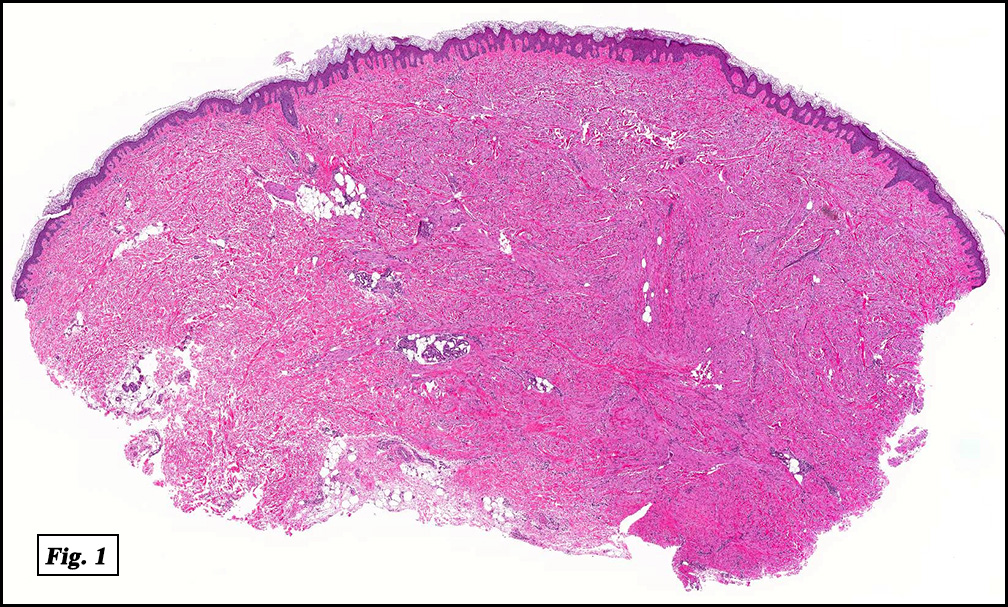
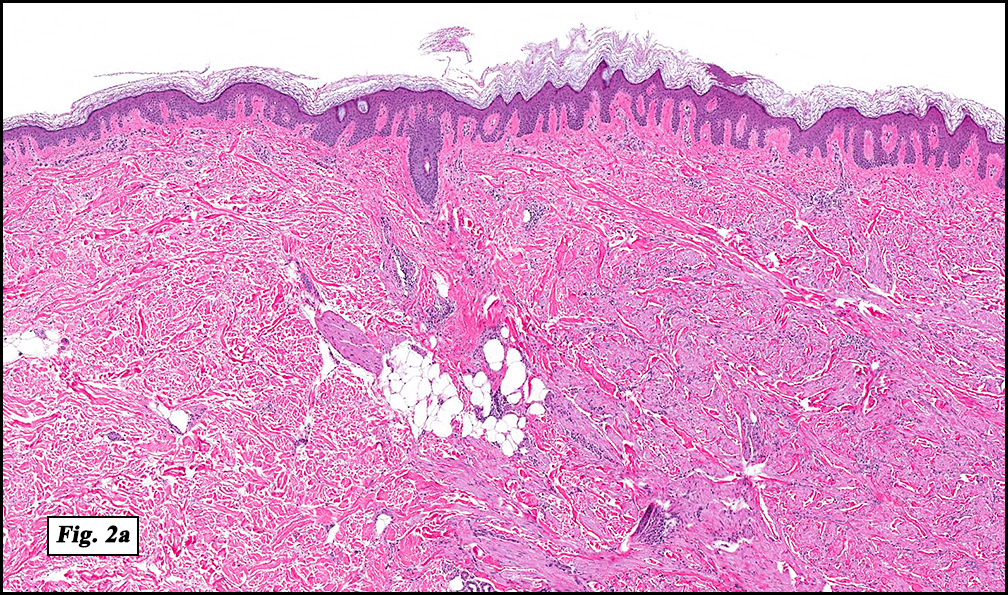
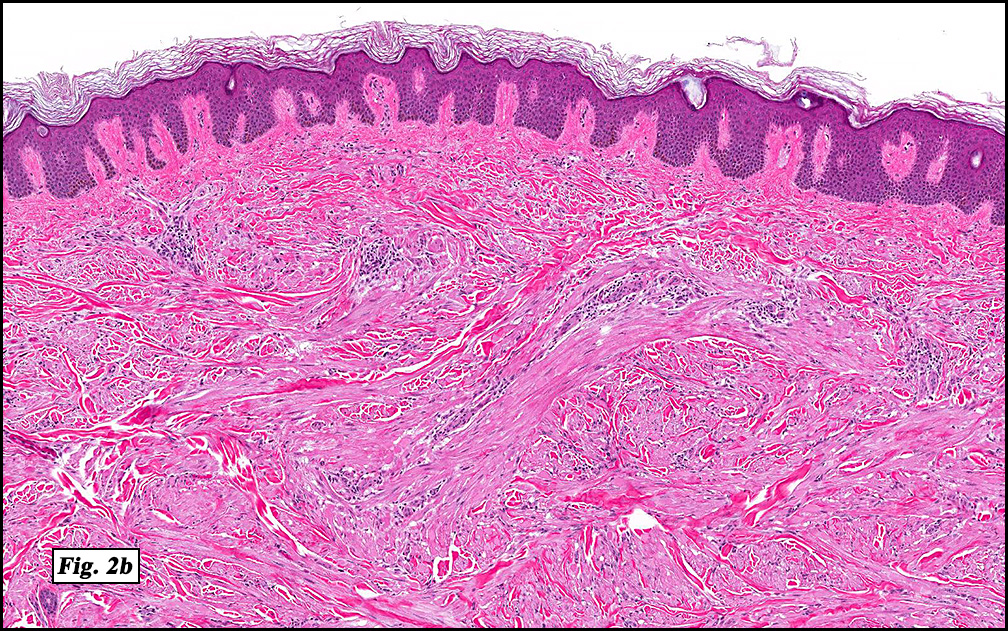
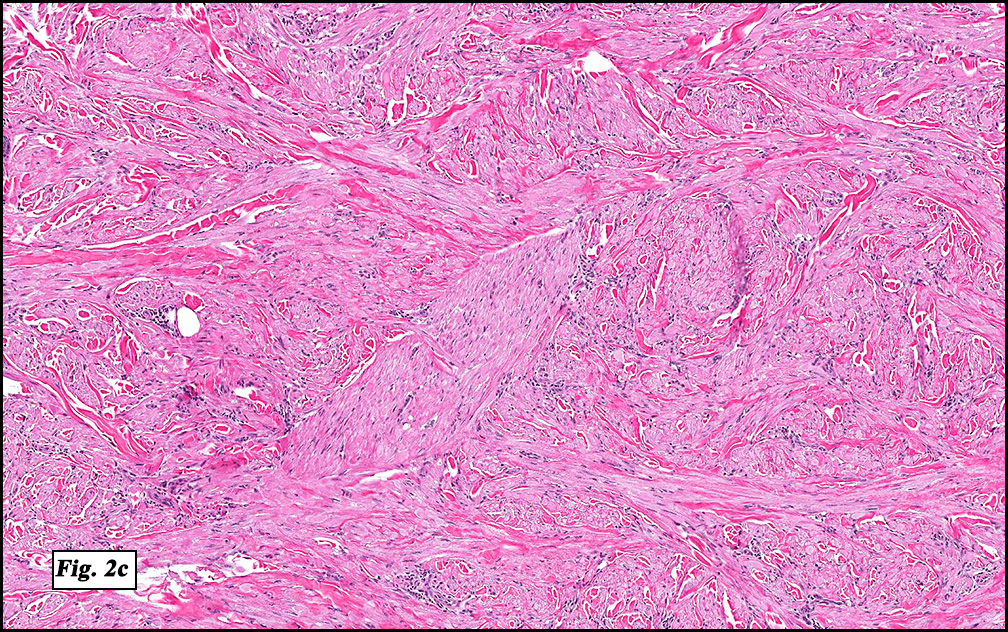
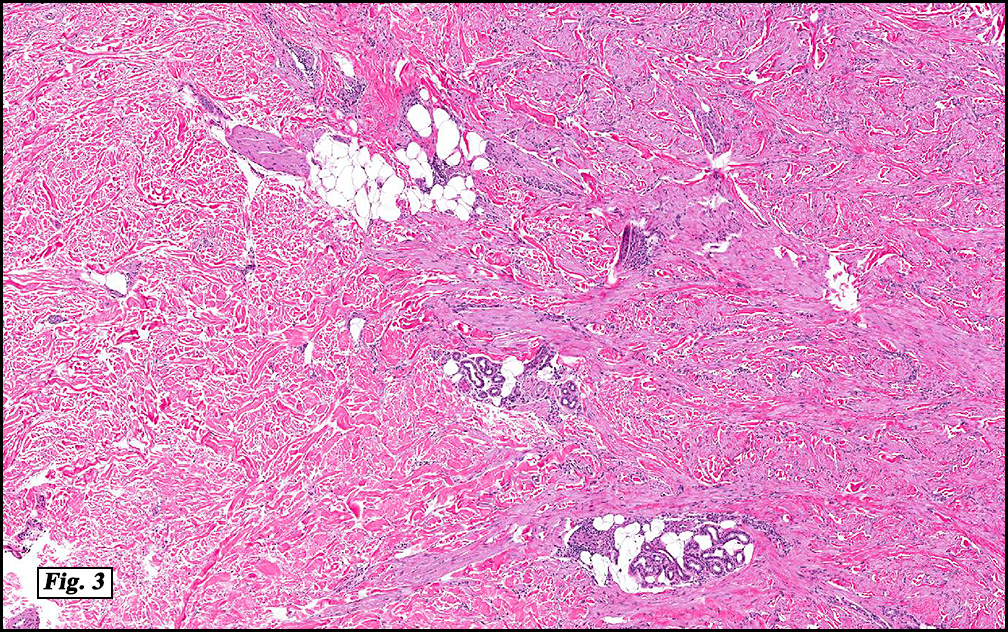
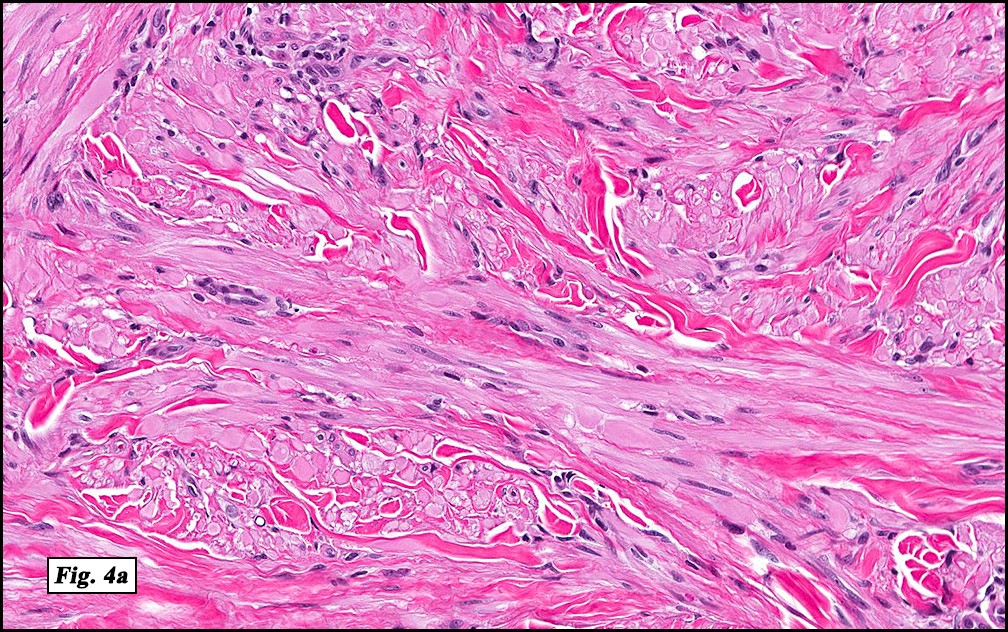
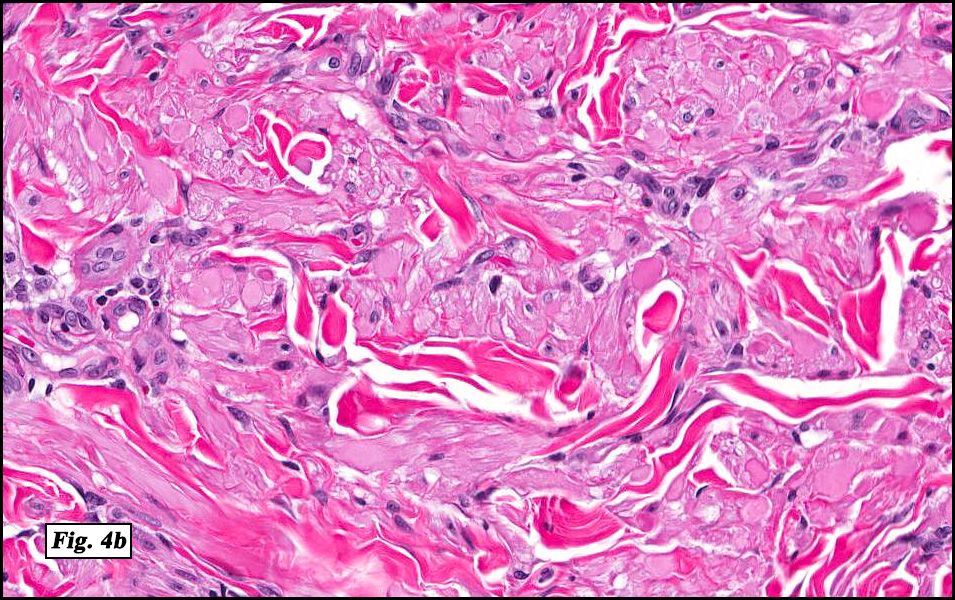
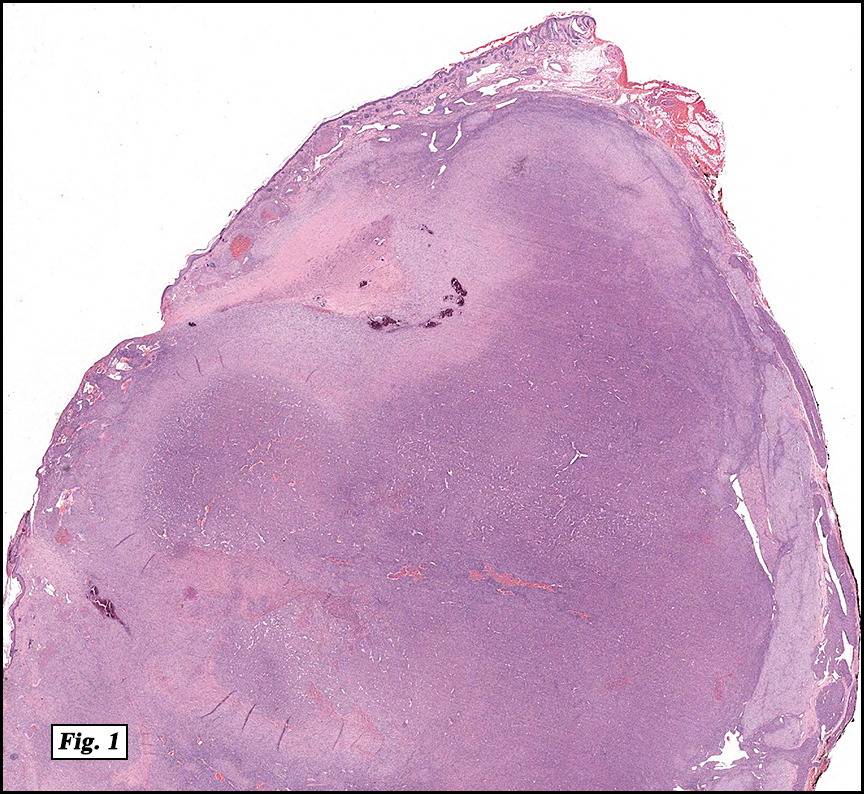
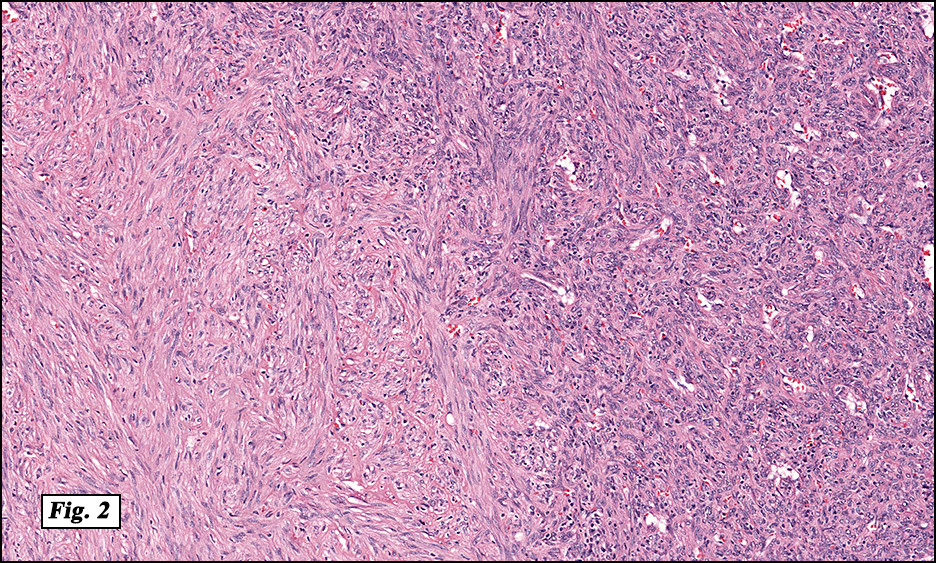
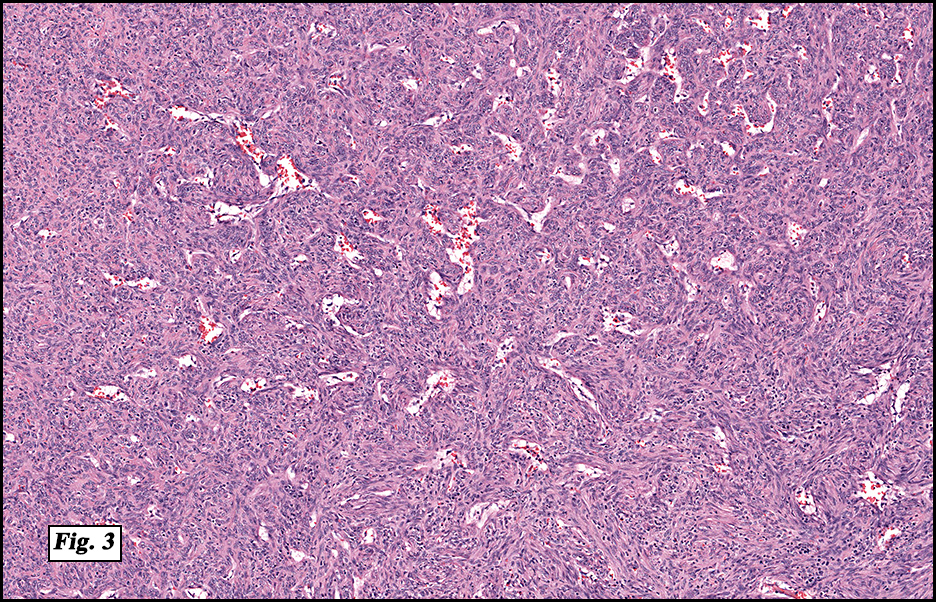
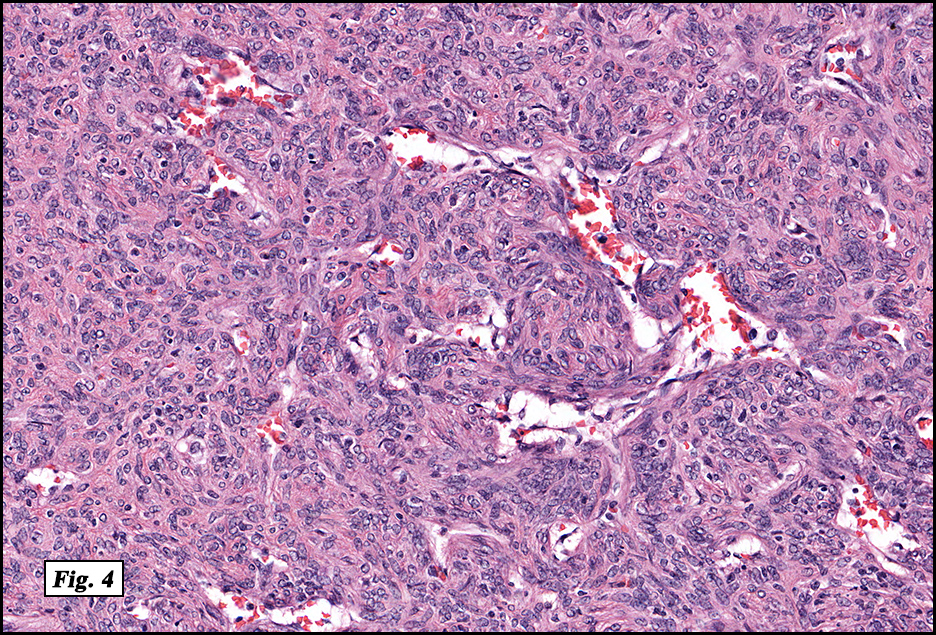
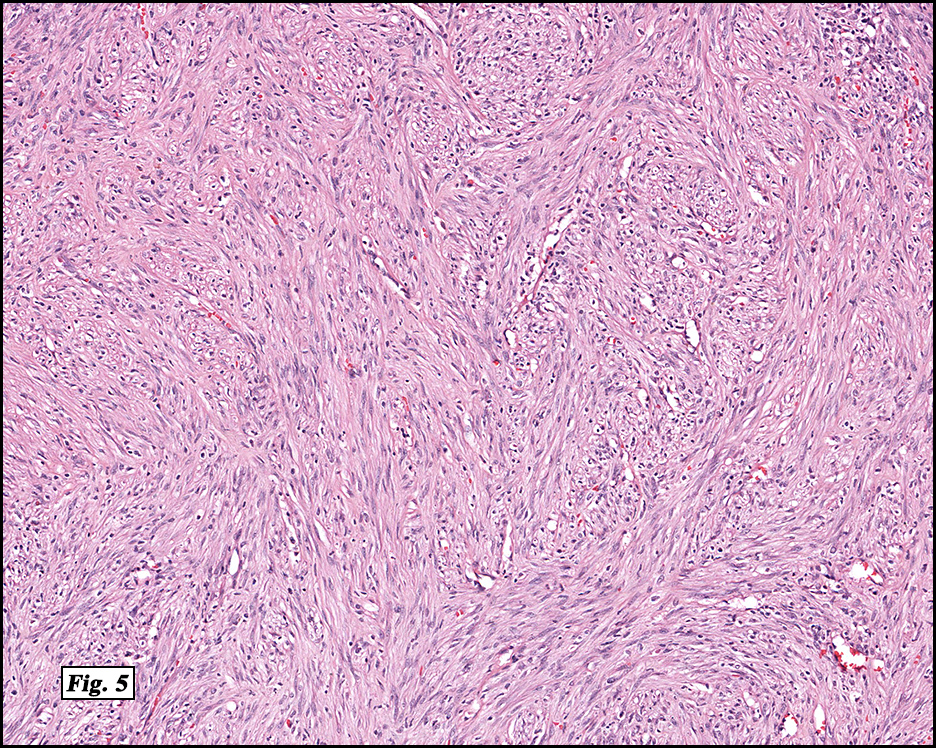
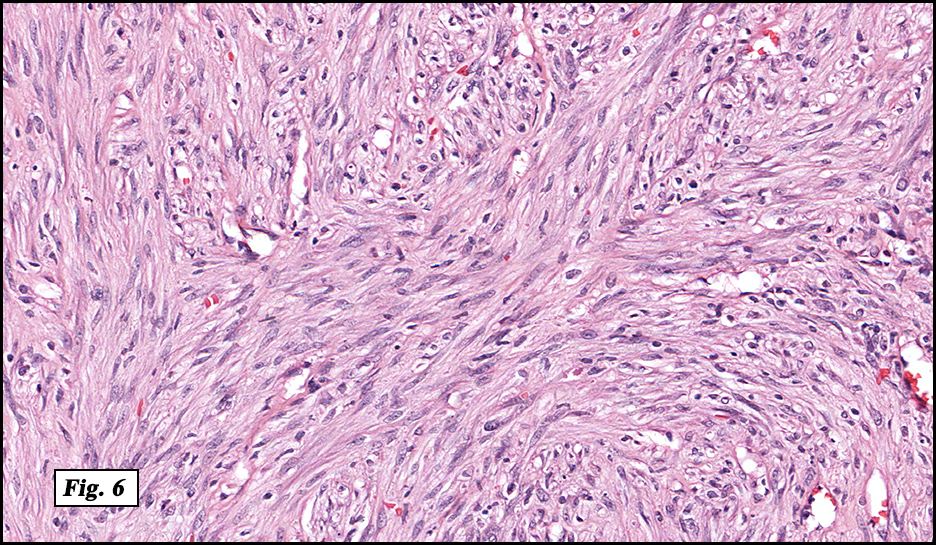
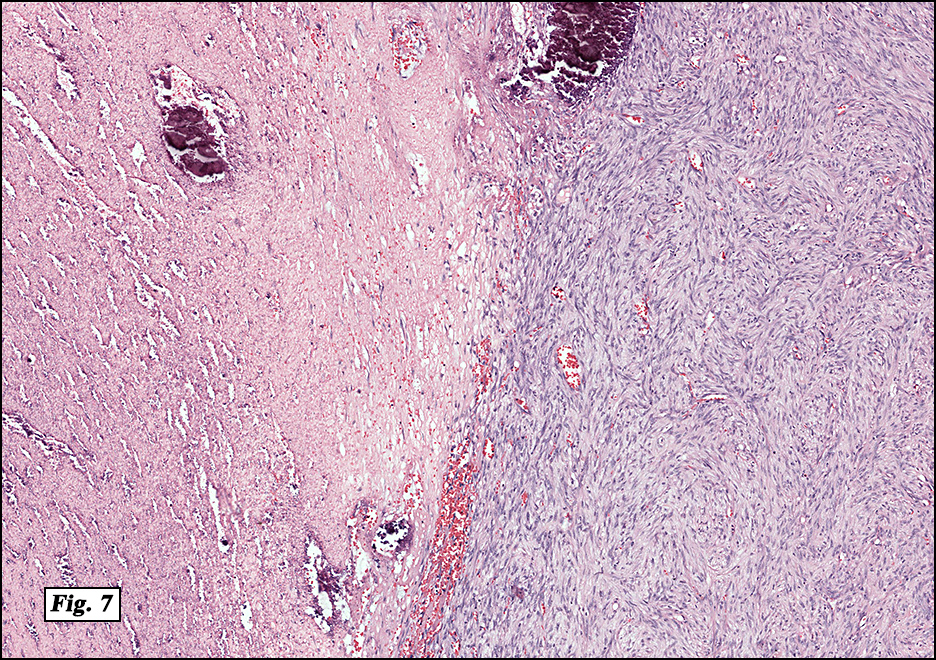
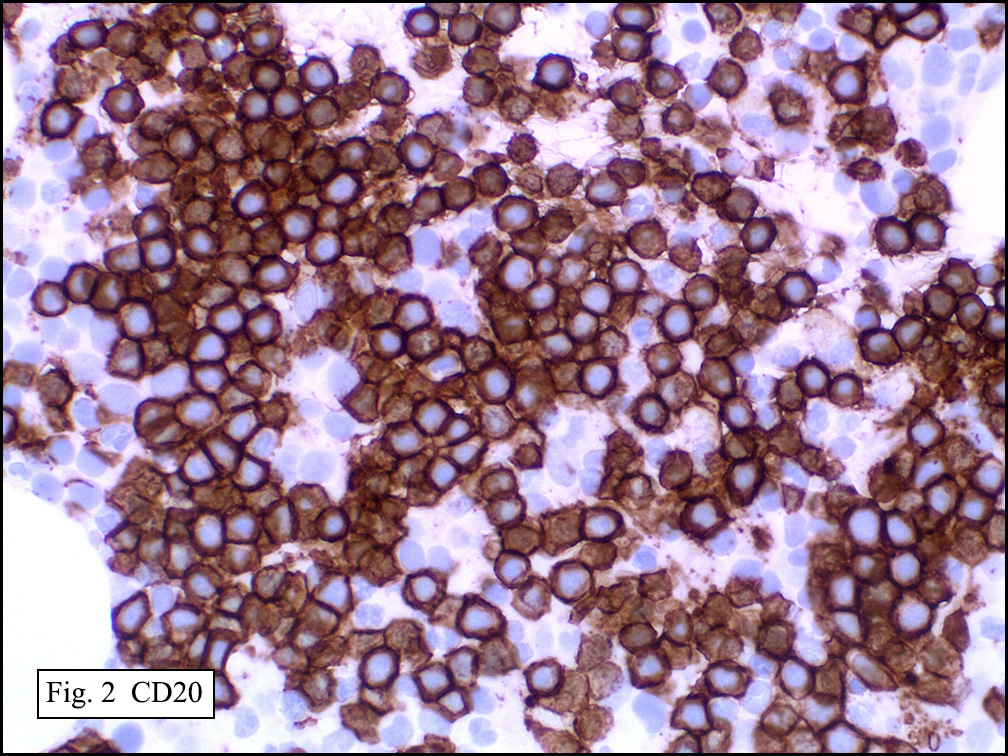
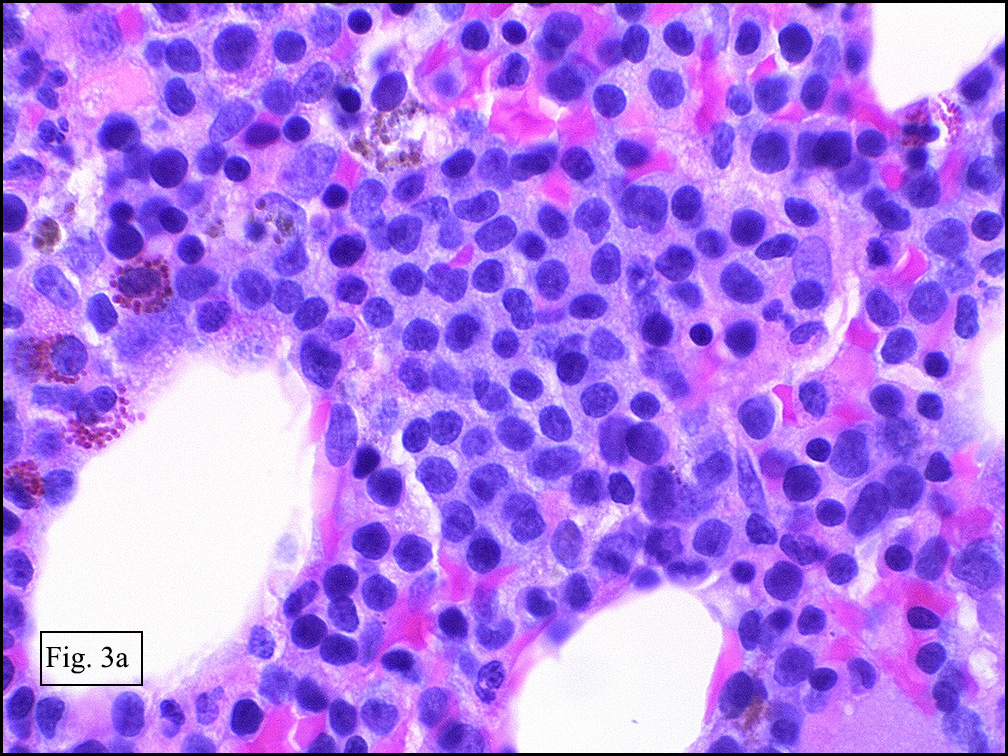
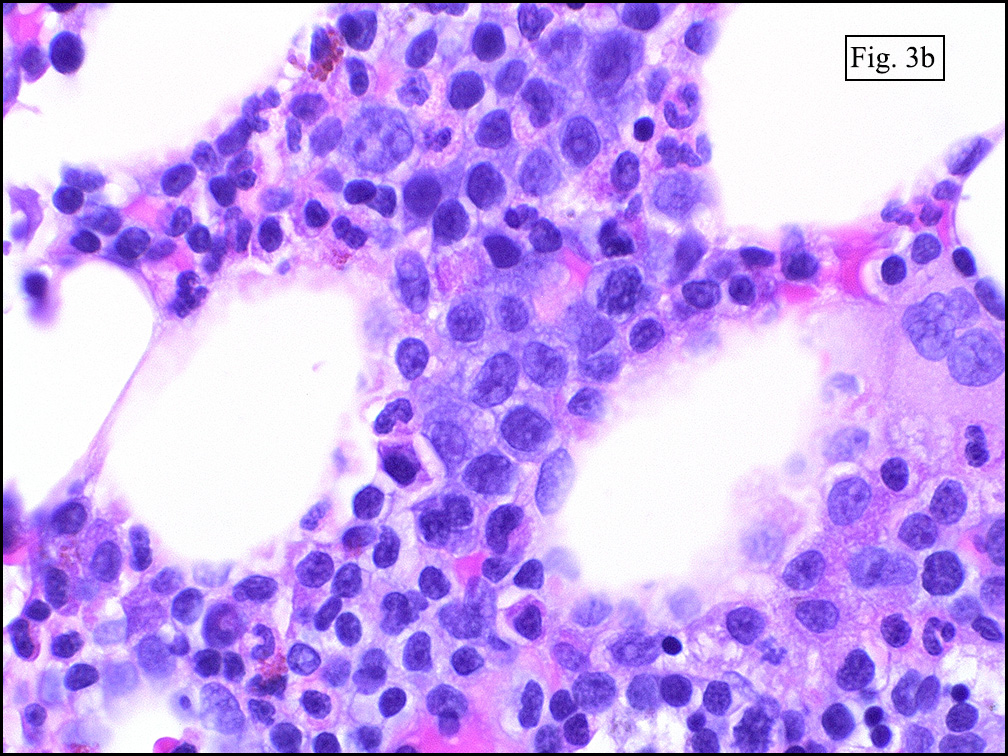
Microscopically, the mass was poorly circumscribed and cellular, located in the dermis and infiltrating skeletal muscle and skin adnexa (Fig. 1). It showed strands and nests of plump cells with eosinophilic cytoplasm in a myxoid background (Fig. 2). In some areas, blood vessel-like structures were present (Fig. 3). Under high amplification these cells had variable-sized vesicular nuclei, prominent nucleoli, cytoplasmic vacuoles and eosinophilic cytoplasm. In most cells the cytoplasmic vacuoles were so prominent that they looked like they had blistered the cells (Fig. 4a, 4b, 4c). Even though nuclear pleomorphism was notable, no atypia or mitotic activity was identified. Immunohistochemical stains showed the tumor cells to be focally positive for factor VIII, but negative for keratin and vimentin.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Diagnosis: Epithelioid hemangioendothelioma
Bingbing Song, M.D. and Donald R. Chase, M. D.
Department of Pathology & Human Anatomy
Loma Linda University Medical Center,
Loma Linda, California
Discussion: Epithelioid hemangioendothelioma (EH) is an angiocentric vascular tumor with metastatic potential and gets the WHO classification as an intermediate grade tumor. It is usually composed of epithelioid endothelial cells with prominent intracytoplasmic vacuoles, arranged in short cords and nests and set in a distinctive myxohyaline stroma.
EH is a rare tumor which may occur in any age group, in either gender and involve virtually any site. It usually presents as a solitary, slightly painful nodule. Deeply situated soft tissue sites may show focal ossification. When EH arises in organs, such as liver and lung, there is a female predilection, possibly due to oral contraceptives.
As original described, EH may arise within or in close approximation to medium-large sized blood vessels. It may mimic an organizing thrombus. Grossly, these tumors have a variegated, white-red color and superficially resemble an organizing thrombus. Some tumors arise from soft tissue instead of blood vessel, but in both types the tumors are composed of short strands or solid nests of round to slightly spindled polygonal endothelial cells. Rarely are large, distinct vascular channels seen, except in the more peripheral regions of the tumor. The tumor cells form small intracytoplasmic lumens, which are seen as clear spaces, or “vacuoles” which can distort or blister the cells. The stroma is usually myxoid or hyaline. Metaplastic bone is occasionally present within large deep lesions and some cases contain prominent osteoclastic giant cells.
Even though most of the tumors appear bland and have no mitotic activity, some tumors contain areas with significant atypia, mitotic activity (>1/10 high power fields), focal spindling of the cells, and/or necrosis. Such features may be related to more aggressive course and poor prognosis. High mitotic figures and tumor size >3.0 cm are associated with a worse prognosis.
The tumor cells are usually positive for CD31, CD34, factor VIII and FLI-1. Focal cytokeratin expression is present in about 25-30%. By ultrastructure, the neoplastic cells are situated on a basal lamina, and possess surface-oriented pinocytotic vesicles and occasional Weibel-Palade bodies.
Genetically, a reciprocal translocation of chromosomes 1 and 3 t(1;3)(p36.3;q25), has been reported.
For the tumors in the soft tissue, the local recurrence rate is 10-15%, the metastatic rate is 20-30%, and mortality is 10-20%. The tumor has more aggressive behavior when it involves visceral organs.
Differential diagnosis includes a variety of vascular tumors and other tumors with epithelioid features in the soft tissue:
• Melanomas and metastatic carcinomas to the soft tissue usually display far more nuclear atypia and mitotic activity than epithelioid hemangioendothelioma.
• Epithelioid hemangioma is composed of multicellular vascular channels which are usually absent in epithelioid hemangioendothelioma and prominent eosinophil infiltration. Mitotic figures are absent.
• Epithelioid angiosarcoma is composed of anastomosing channels lined by highly atypical, mitotically active epithelioid endothelial cells. Necrosis is common.
• Epithelioid sarcoma consists of nodules of rounded eosinophilic cells that surround cores of necrotic debris and collagen. Epithelioid sarcoma usually arises in distal extremity, particularly hand, in the young individuals. The tumor cells are positive for cytokeratin, CD34 (50%), EMA and may be positive for CD31. They are negative for factor VIII and FLI-1.
In summary, epithelioid hemangioendothelioma is an intermediate grade, usually angiocentric vascular tumor with metastatic potential. It can be found in both soft tissue and organs. It is composed of epithelioid endothelial cells with prominent intracytoplasmic vacuoles, arranged in short cords and nests in a distinctive myxohyaline stroma and usually marks for vascular markers.
Suggested Reading:
Fletcher CDM, Unni KK, Mertens F, et al. World Health Organization Classification of Tumor, Pathology and Genetics Tumors of Soft Tissue and Bones. 2002.
Weiss SW, Goldblum JR, et al. Enzinger and Weiss’ Soft Tissue Tumors. 5th edition. 2008.
Kempson RL, Fletcher CDM, Evan HL, et al. Atlas of Tumor Pathology, Tumors of Soft Tissue. 3rd series, 1998.
Mentzel T, Beham A, Fletcher CDM, et al. Epithelioid hemangioendothelioma of skin and soft tissues: clinicopathologic and immunohistochemical study of 30 cases. Am J Surg Pathol.1997 Apr 21(4):363-74.
Deyrup AT, Tighiouart M, Weiss SW, et al. Epithelioid hemangioendothelioma of soft tissue: a proposal for risk stratification based on 49 cases. Am J Surg Pathol. 2008 Jun 32(6):924-7.
Radin DR, Craig JR, Halls JM, et al. Hepatic epithelioid hemangioendothelioma. Radiology 1988 Oct;169(1):145-8.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}