History: 74-year-old gravida 1, para 1 female with a history of hysterectomy and bilateral salpingoopherectomy for severe pelvic endometriosis who presented with abdominal pain. A CT scan demonstrated a 6 cm pelvic mass lying between the bladder and rectum. Fecal occult blood test was positive. The patient underwent attempted resection of the pelvic mass, as well as a sigmoid colon resection.
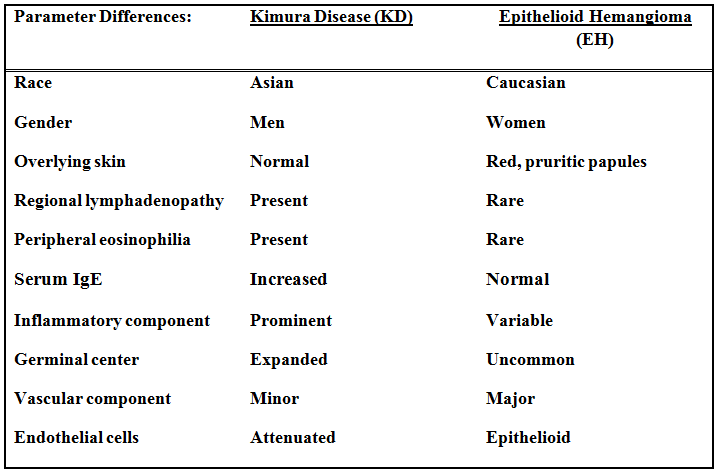
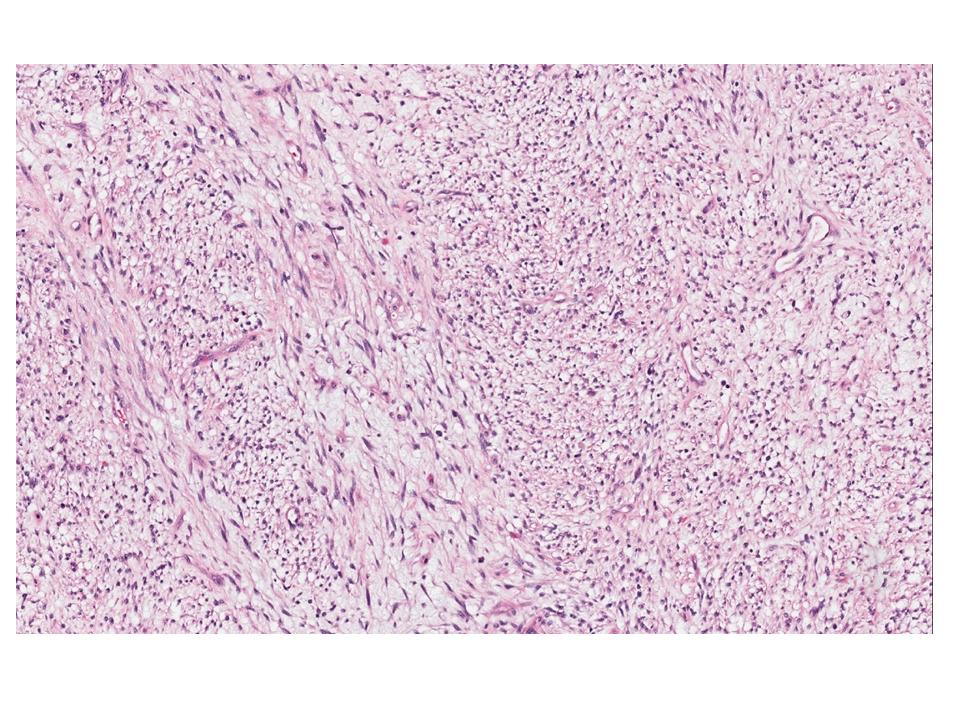
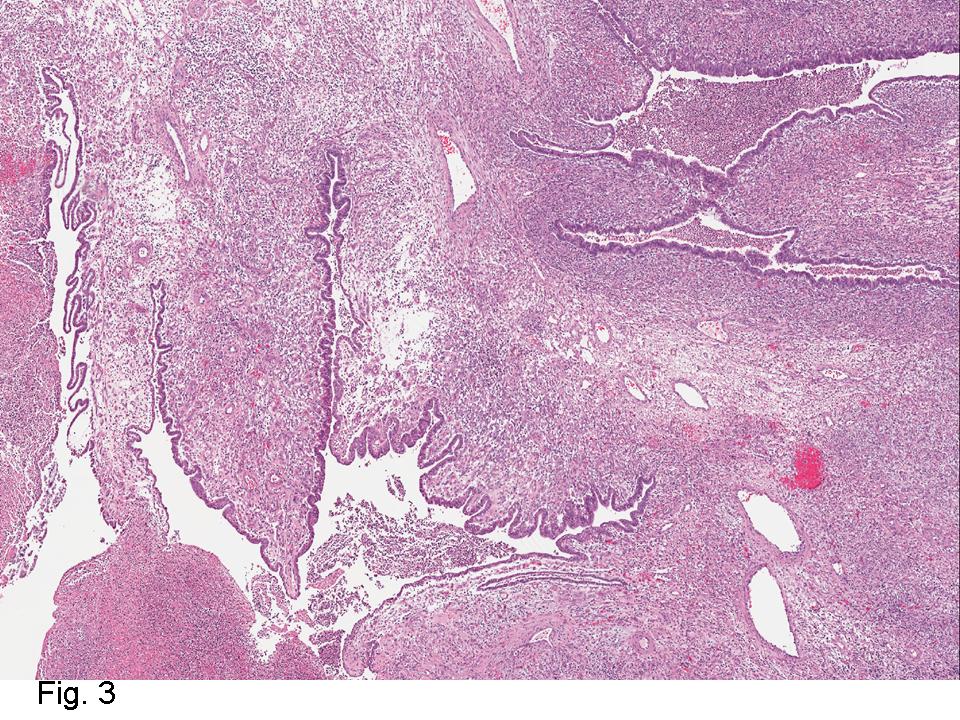
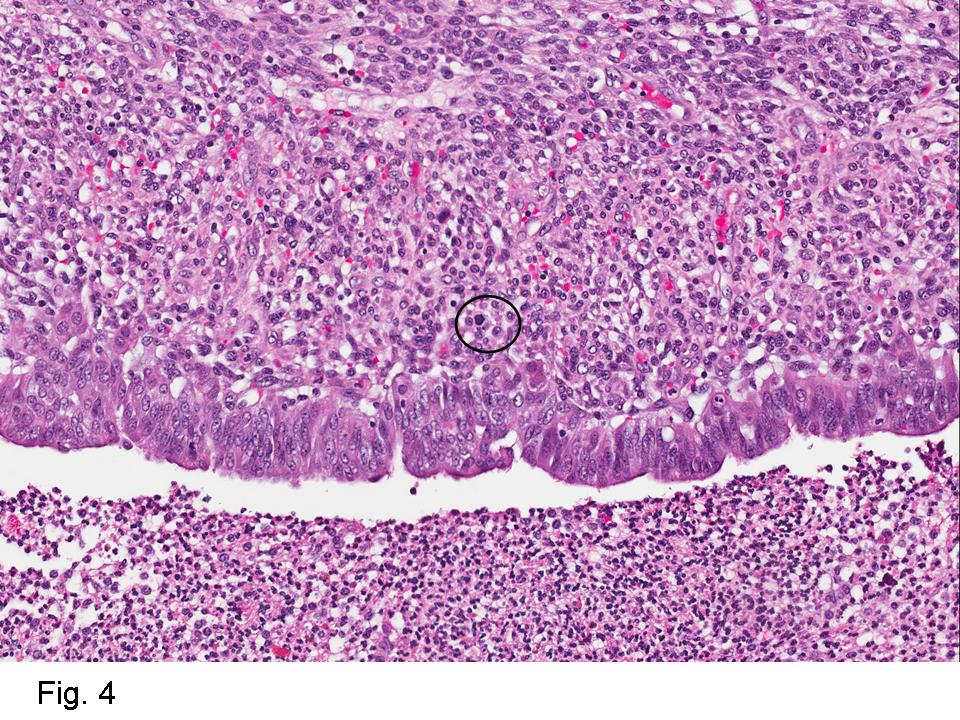
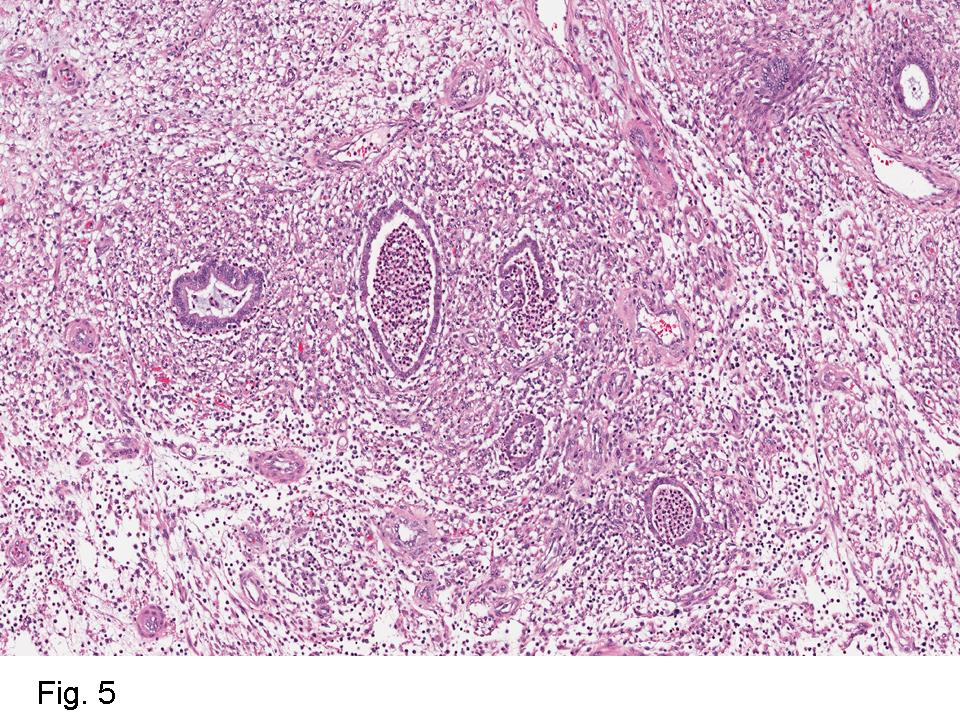
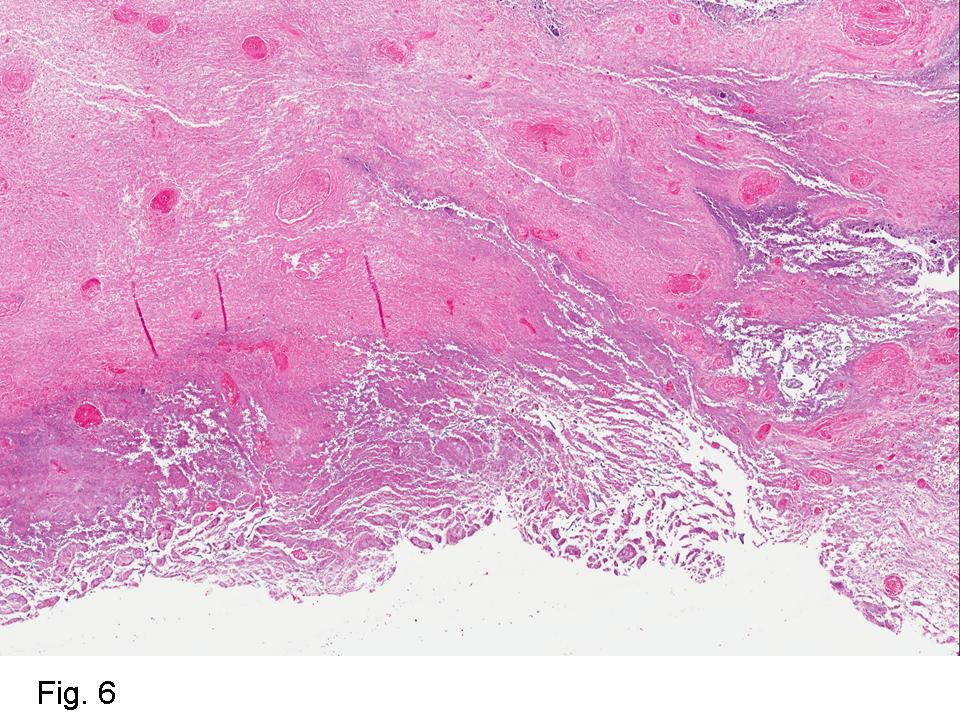
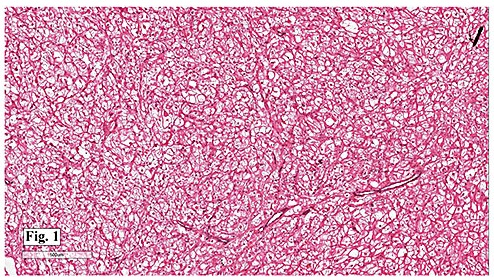
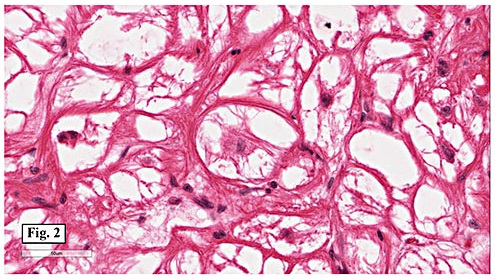
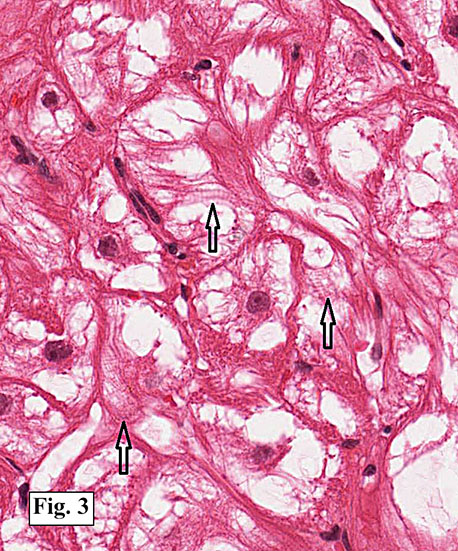
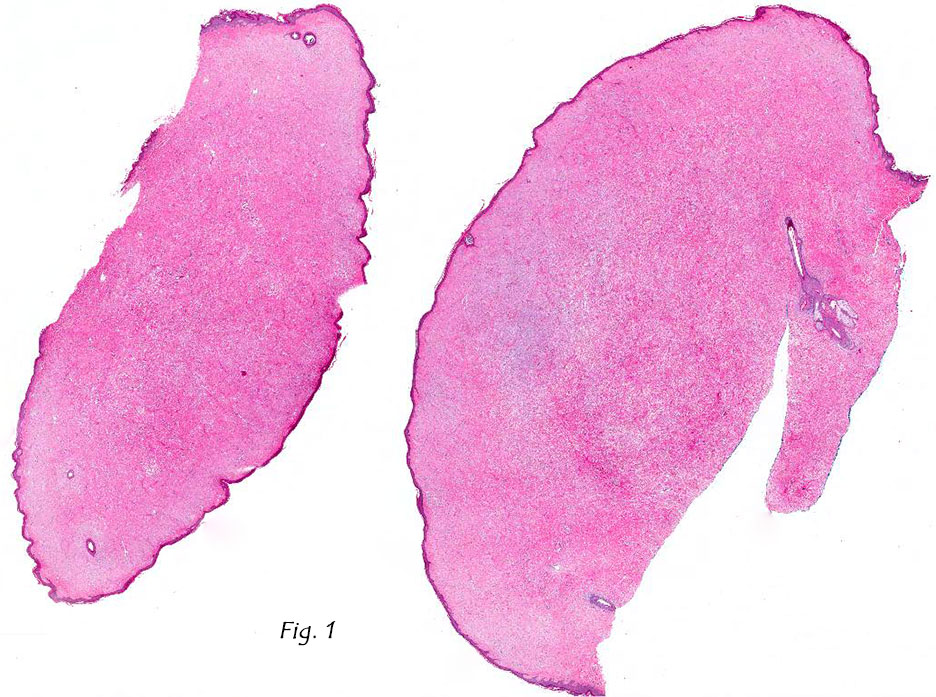
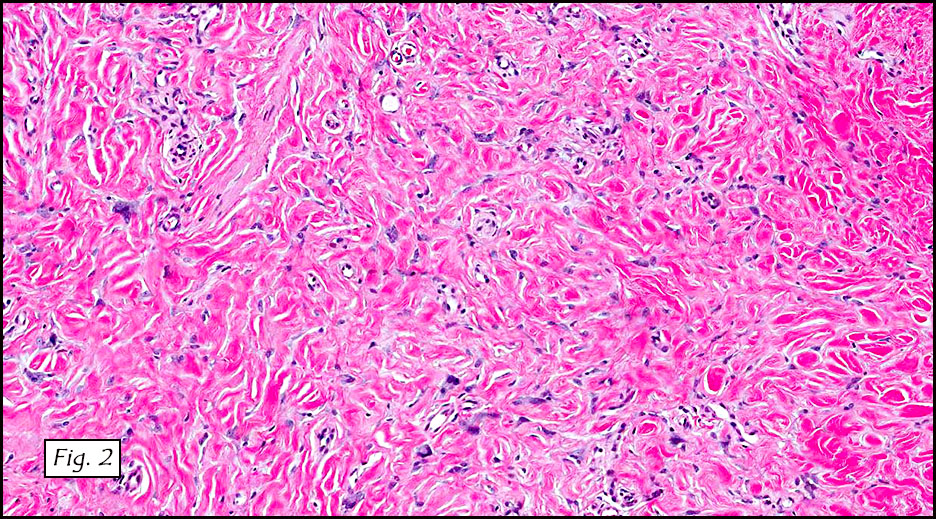
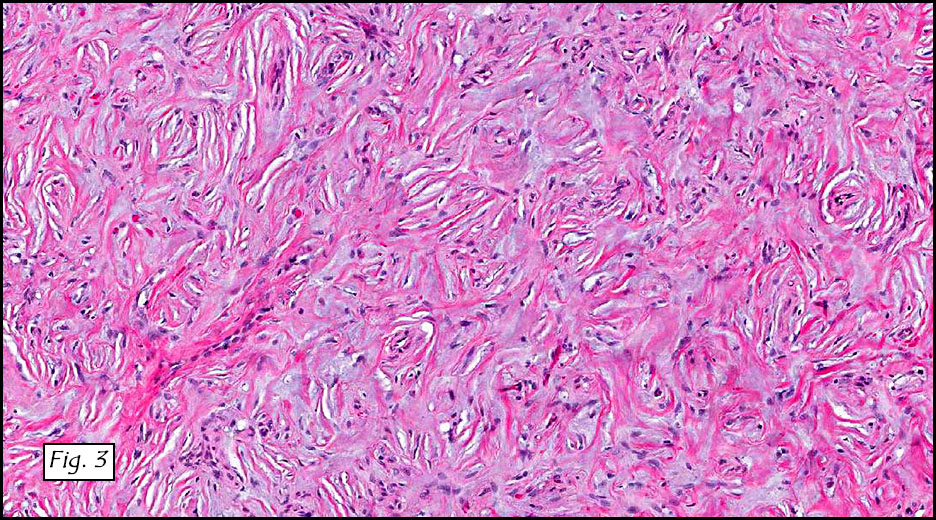
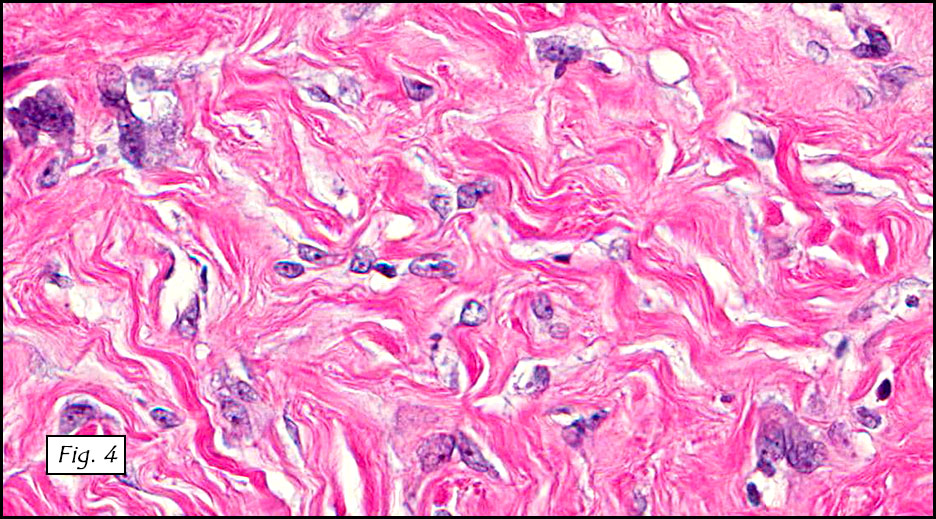
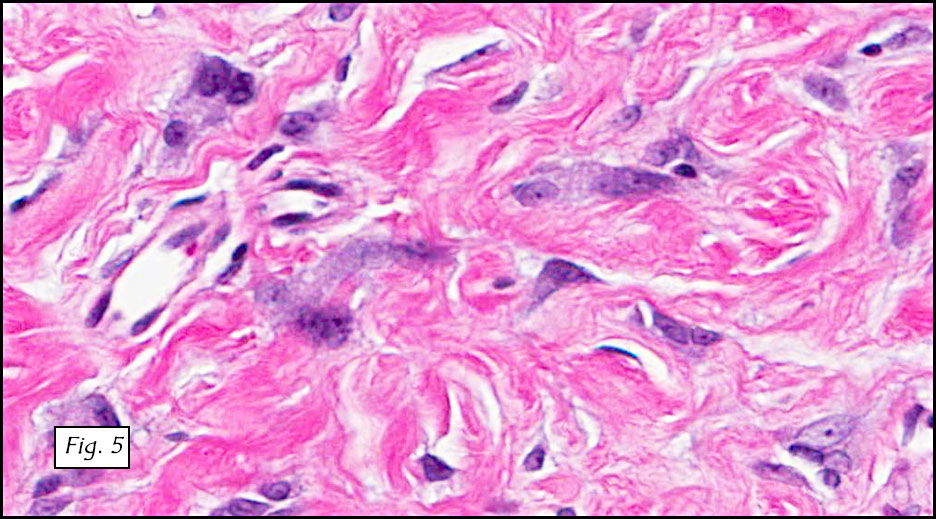
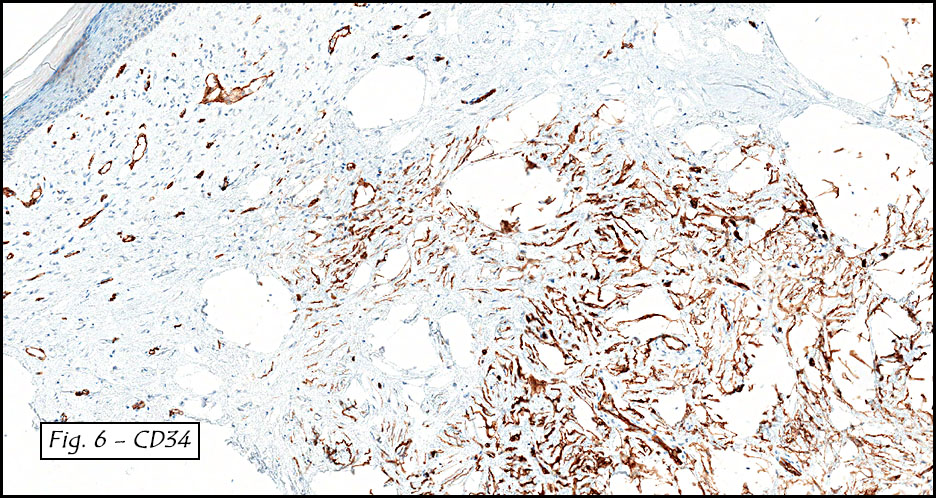
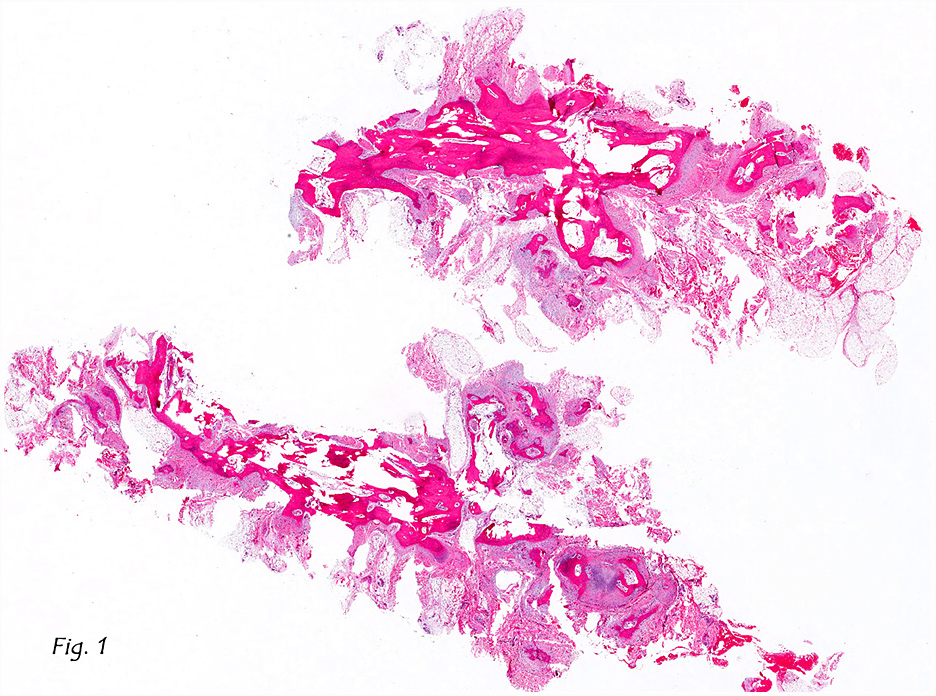
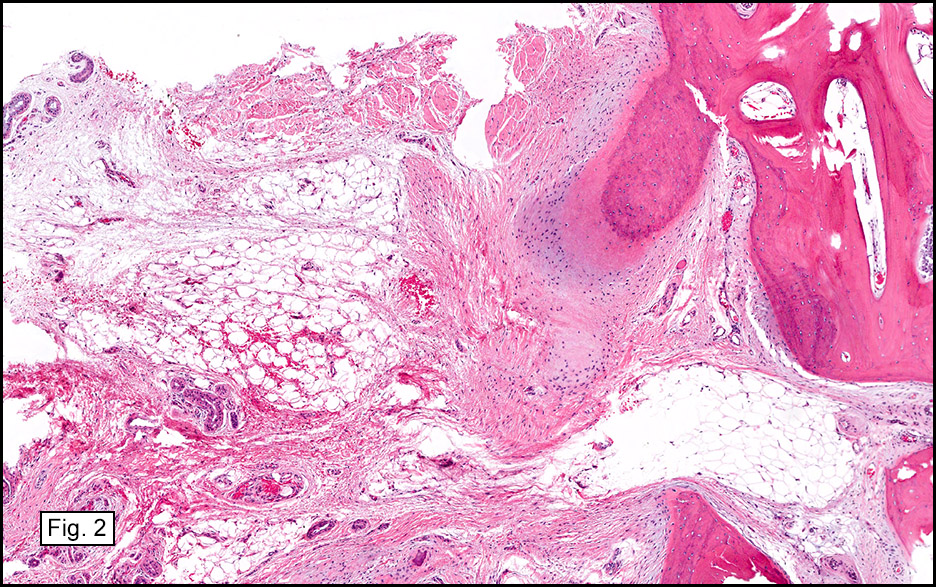
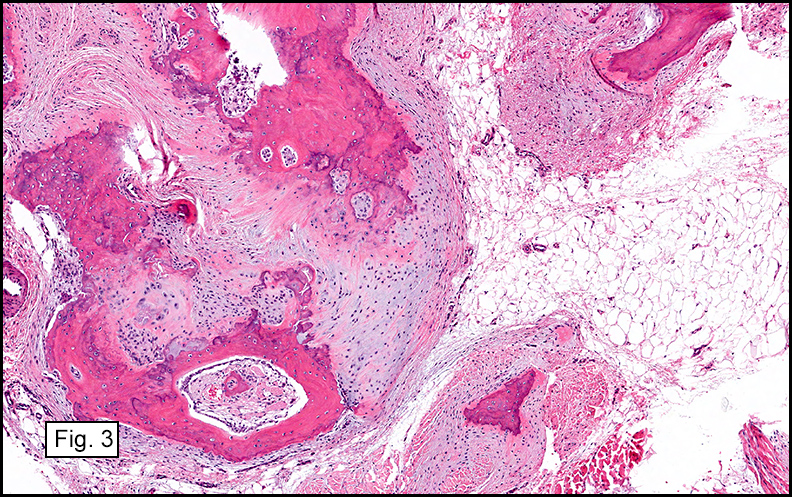
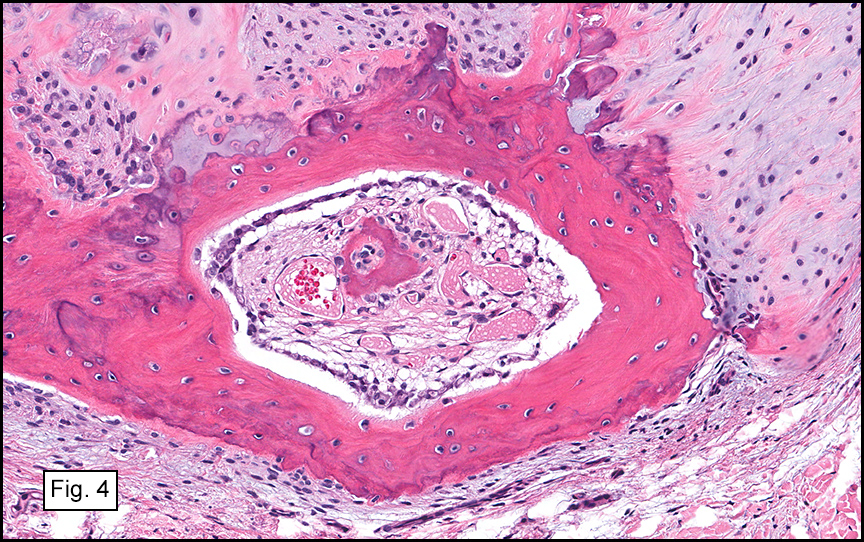
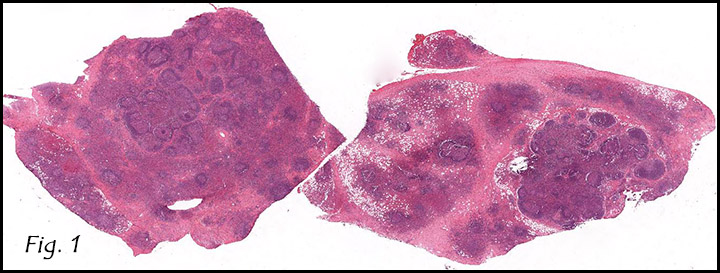
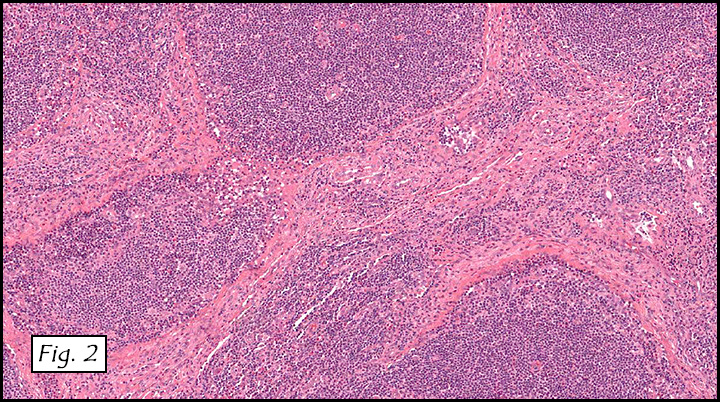
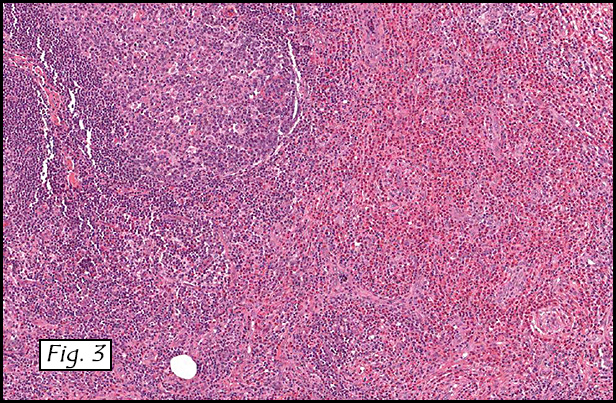
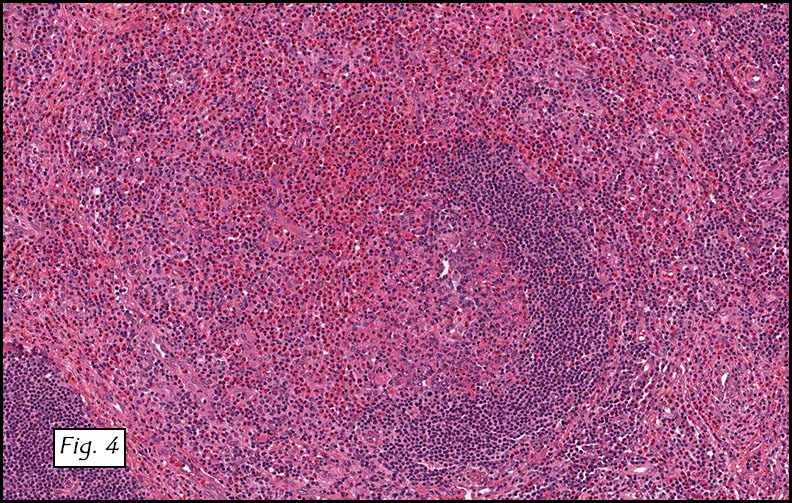
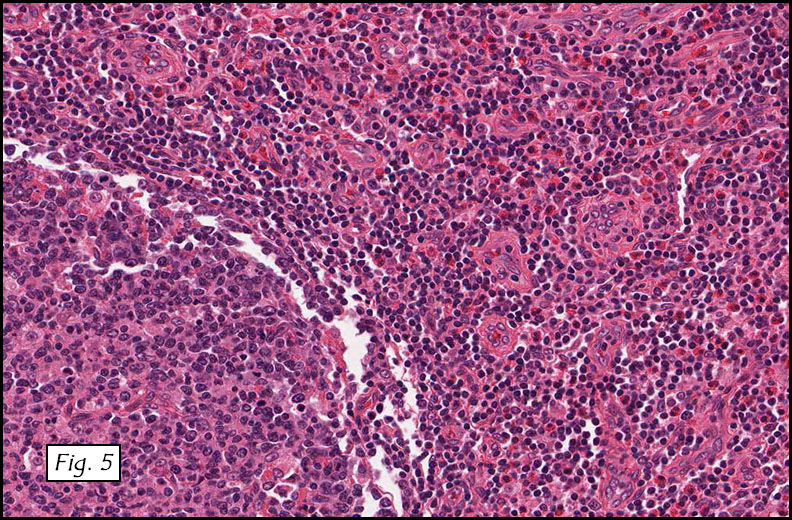
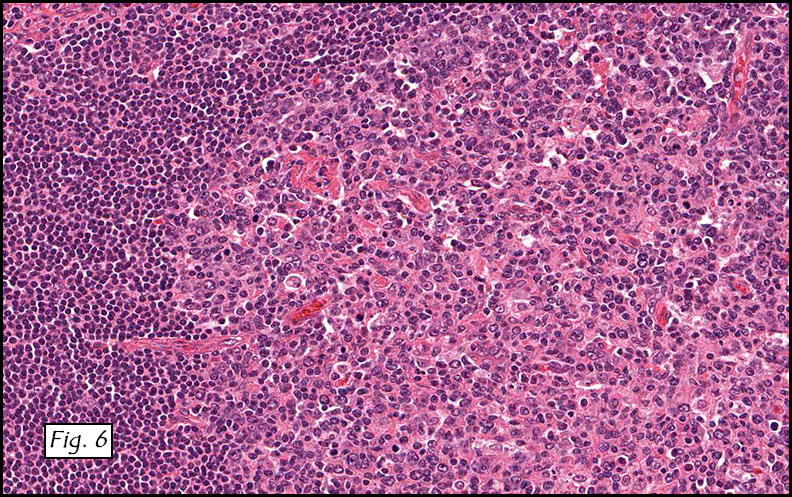
Gross examination of the resection specimens demonstrated a 10 cm mass that is adherent to the sigmoid colon mass. Also received was a 3 cm aggregate of fragments of tumor designated as pelvic tumor. Microscopically, the tumor comprised large expanses of a spindled mesenchymal neoplasm with relatively monotonous nuclear features, moderate atypia, and indistinct cytoplasm (Fig.1). Occasional areas showing true epithelial gland formation were present, with surrounding hypercellular condensations of the atypical mesenchymal elements (Fig.2). There was prominent stromal overgrowth, resulted in polypoid epithelial projections forming “leaf-like’ low-power appearance (Fig.3). Mitotic figures were easily identified (up to 8 per 10 HPF) (Fig.4). Foci of endometriosis in the bowel serosa were also present (Fig.5). The overlying bowel mucosa was ulcerated, but was without dysplasia (Fig.6).
Diagnosis:
Extrauterine Mullerian Adenosarcoma with Sarcomatous overgrowth
Suneetha Chintalapati, M.D., Donald R. Chase, M.D. and Jeremy K Deisch, M.D.
Department of Pathology and Human Anatomy
Loma Linda University Medical Center, Loma Linda, California
California Tumor Tissue Registry, Loma Linda, California
Discussion: Mullerian Adenosarcoma is an uncommon mixed epithelial-mesenchymal tumor of low-grade malignancy that affects the female genital tract. Mullerian adenosarcoma is regarded as a lesion with intermediate pathologic features and prognosis, lying between benign adenofibroma and carcinosarcoma.
Extrauterine Mullerian adenosarcoma is very rare, and is presumed to arise in foci of endometriosis, given its frequent co-existence (as seen in this case). Extrauterine examples are more aggressive than uterine tumors, due to ease of access to the peritoneal cavity, as it is not retarded by the thick myometrial wall. The recurrence rate following surgery is higher. Extrauterine Mullerian adenosarcomas are the second most common malignancy arising in extraovarian endometriosis, following clear cell carcinoma. Exogenous or endogenous estrogen may have some role in the transformation process, whereas the effectiveness of combined estrogen–progesterone hormone replacement therapy is still to be determined.
There are currently two recognized sets of criteria for the pathologic diagnosis of malignant transformation of endometriosis. Sampson’s criteria for malignant transformation of endometriosis requires that there is a focus of endometriosis in close proximity to malignancy, that the tumor shows a histological appearance compatible with endometrial origin, and that that other primaries are appropriately excluded. The criteria set forth by Clement and Scully requires a mitotic count of two or more per ten high-power fields, the presence of stromal cellular periglandular cuffs, intraglandular protrusion of cellular stroma, and at least a moderate degree of nuclear atypia of the mesenchymal elements.
Pathologically, Mullerian adenosarcomas are biphasic tumors, in which there are glands lined by benign or mildly atypical Mullerian-type epithelium, intimately mixed with a proliferating mesenchymal component that is a low grade sarcoma, with cellularity more pronounced at the periphery of the glands, forming periglandular cuffs. The immunoprofile is similar to endometrial stromal sarcoma: CD10, WT1, ER and PR are usually positive.
The immunoreactivity for adenosarcomas is similar to that of adenofibromas, which are associated with favorable outcome, as well as other benign lesions such as endometrial polyps and endometriosis. The significant histologic and immunophenotypic overlap between adenosarcomas and adenofibromas suggest that some of the tumors currently classified as adenofibromas, on the basis of their low mitotic count and lack of significant nuclear atypic, are, in fact, well-differentiated adenosarcomas.
Adenosarcomas with sarcomatous overgrowth differ histologically and prognostically from typical adenosarcomas. Sarcomatous overgrowth requires the presence of stromal predominance be present in at least 25% of the tumor volume. In tumors with sarcomatous overgrowth, the mortality rate is as high as 75%, with a similar malignant potential to high grade uterine sarcomas. These tumors may show a high proliferative index by Ki-67 immunostaining, and may show strong p53 staining and loss of CD10 and progesterone receptor immunoexpression.
Malignant transformation of gastrointestinal endometriosis is rare, but when it occurs, it predominantly affects postmenopausal patients. Recognition of these lesions is important as primary gastrointestinal neoplasms are managed differently than those arising in endometriosis. An adenosarcoma arising from endometriosis should be considered in the differential diagnosis of a pelvic mass, even one appearing in the colon wall, because ectopic endometrial tissue has the potential to exist throughout the peritoneal cavity.
Suggested Reading:
Atlas of Gynecologic Surgical Pathology – Clement, Young, 2014, third edition, 256-259.
Clement PB, Scully RE. Extrauterine mesodermal (mullerian) adenosarcoma: A clinicopathologic analysis of five cases. Am J Clin Pathol 1978;69:276–83.
Mullerian adenosarcoma arising from rectal endometriosis, Chunseok Yang etal, Ann Coloproctol. Oct 2014, 30(5): 232-236.
Mullerian Adenosarcoma of the uterus with sarcomatous overgrowth, reecha singh et al, Clin Med Insights case rep. 2010; 3:27-30.
Neoplastic and Pre-neoplastic changes in gastrointestinal endometriosis; Rhonda k. yantiss metal; Am J Surg Pathol 24(4): 513-524, 2000.
Rosai and Ackerman’s Surgical Pathology, 2011, Tenth edition, Volume 2, 1507-1508.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}