History:Â A 21 year old man presented with an 11 month history of a slow growing, painless subcutaneous mass in the left inguinal area. A pelvic X-ray showed a 7 x 4 x 2.8 cm well circumscribed periarticular calcified mass within the soft tissue of the hip. It consisted of multiple small calcified nodules. Serum calcium, phosphate and alkaline phosphatase levels were normal.
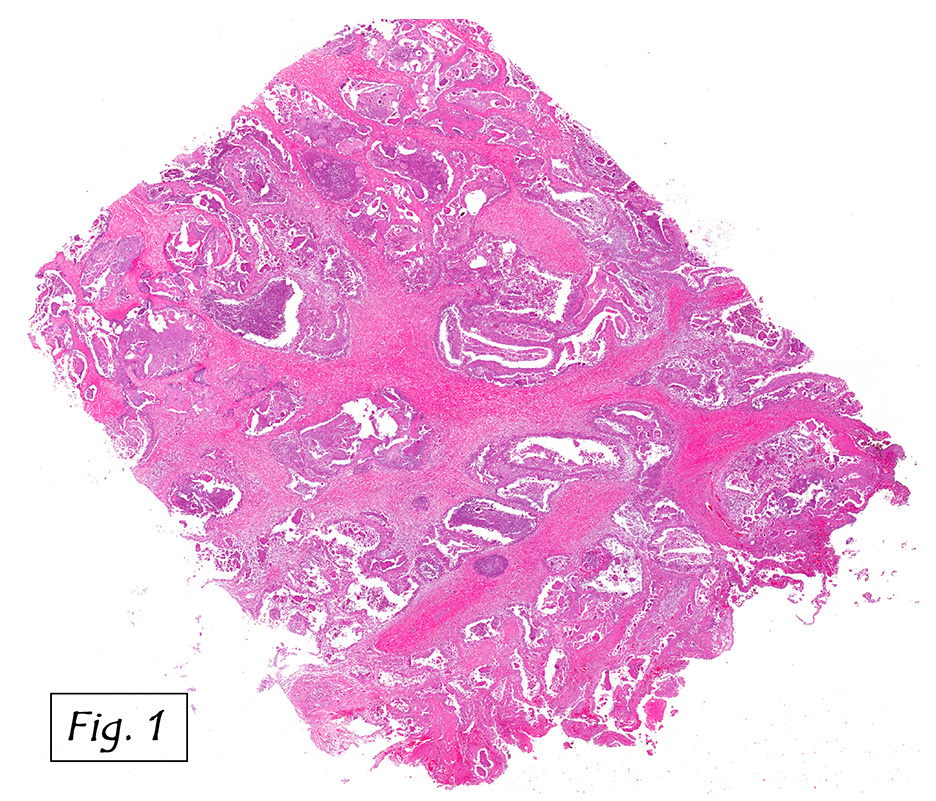
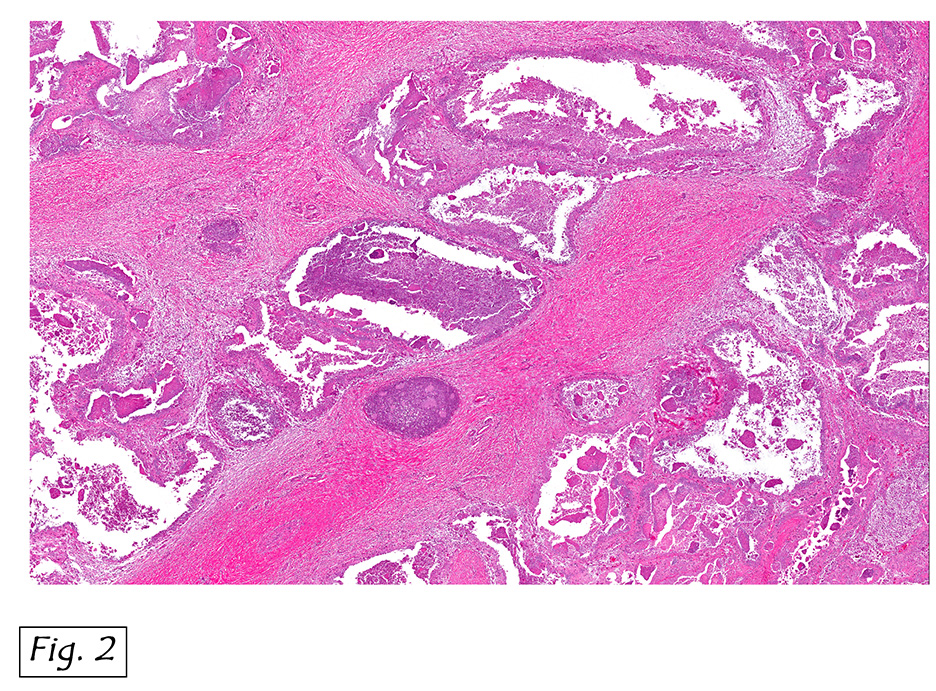
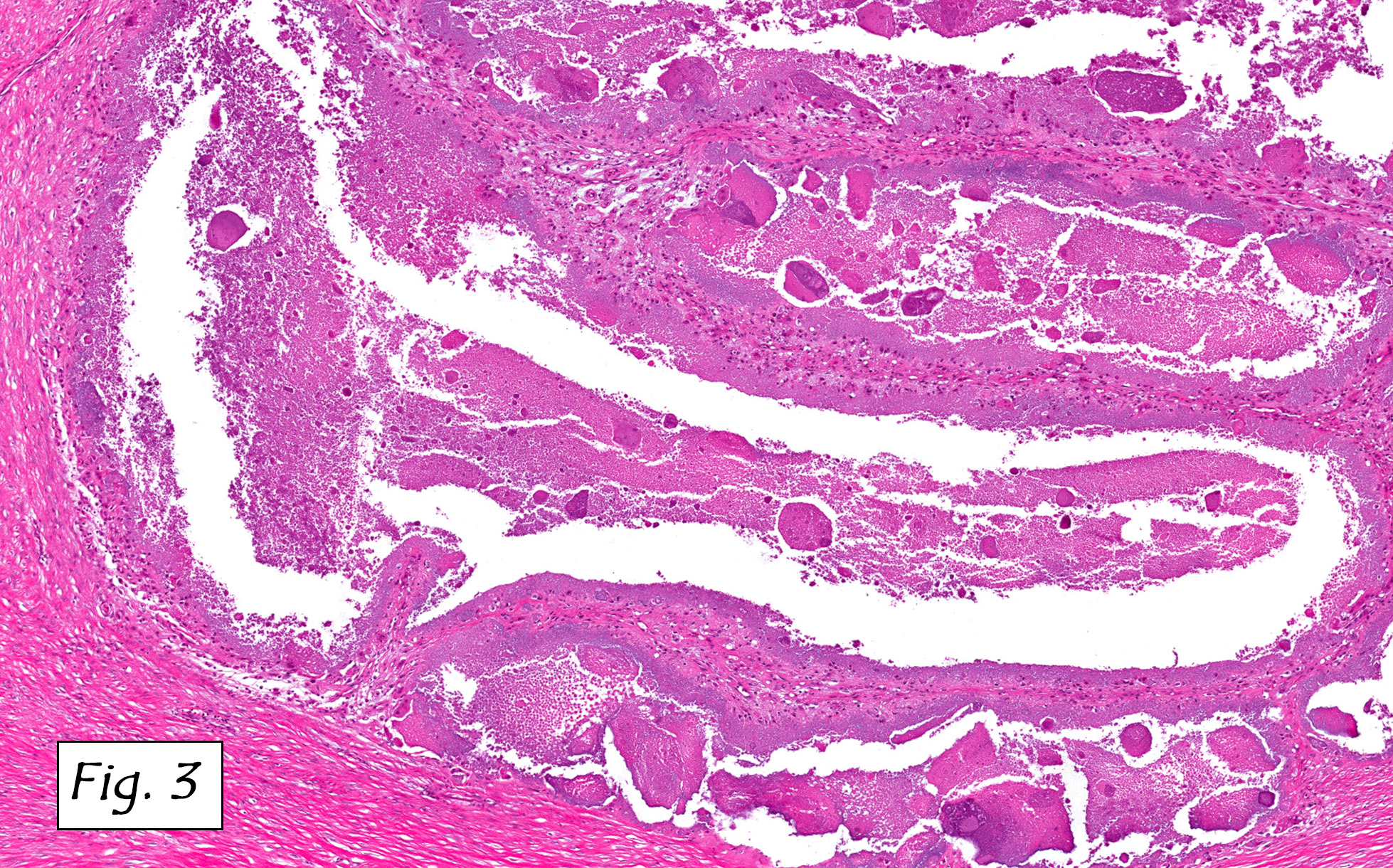
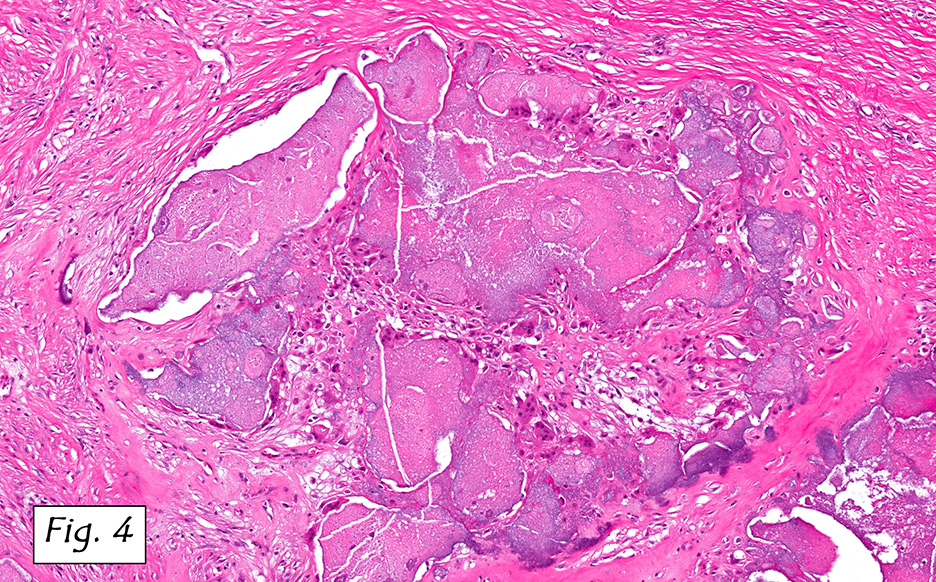
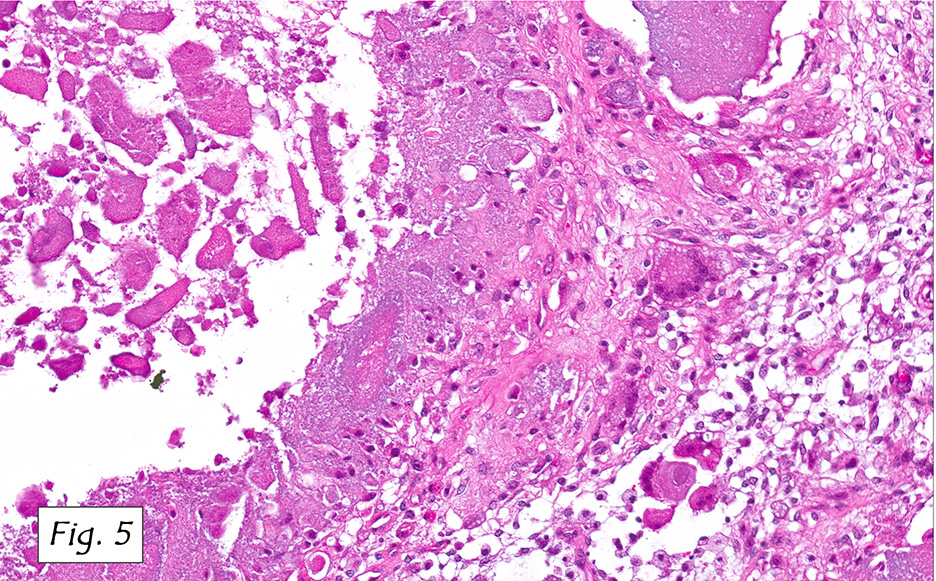
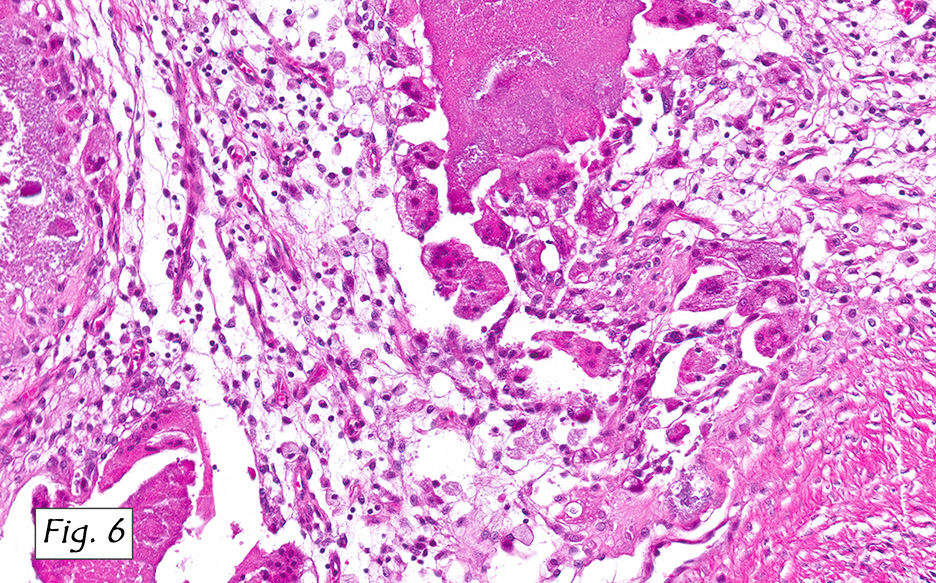
The mass was composed of white, granular material surrounded by a fibrous tissue capsule. The cut surface was hard, calcified, light tan and chalky. Microscopically, it showed multiple, solitary and confluent nodules of calcification in a matrix of granular debris (Figs. 1, 2, 3, 4 ). Associated were histiocytes, multinucleated giant cells and fibroblasts (Figs. 5, 6).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Diagnosis: Tumoral Calcinosis
Huina Zhang, M.D., Ph.D. and Donald R. Chase, M. D.
Department of Pathology and Human Anatomy,
Loma Linda University Medical Center, Loma Linda, California
California Tumor Tissue Registry, Loma Linda, California
Discussion: Tumoral calcinosis is a rare sporadic or familial entity characterized by abnormal peri-articular deposits of calcium hydroxyapatite, most commonly found around major joints, especially the hips, shoulders, and elbows without any abnormalities in the calcium metabolism. It was first described by Duret in 1889 and named by Inclan in 1943. These lesions are predominantly seen during the first two decades of life and are often multicentric. It occurs as a well-defined pathologic entity in three heterologous groups of diseases– sporadic tumoral calcinosis, familial hyperphosphatemic and normophosphatemic tumoral calcinosis.
• The sporadic form presents with a solitary, large, firm subcutaneous calcified mass around large joints, often attached to tendon or deep fascia without involving the underlying bone and joints.
• The familial hyperphosphatemic and normophosphatemic tumoral calcinosis forms are extremely rare and transmitted in an autosomal recessive manner with variable clinical expression. Germline mutations in the GALNT3, FGF23, KL or SAMD9 gene have been recently described in familial cases. Imaging studies reveal a subcutaneous conglomerate of multiple, rounded opacities separated by radiolucent lines imparting a chicken-wire pattern of lucencies with distinct fluid levels in some of the nodules. Due to a high recurrence rate, treatment is complete surgical excision.
Grossly, tumoral calcinosis is a poorly-defined, firm, rubbery mass, ranging in size between 5-15 centimeters. Cut surfaces demonstrate multilocular cystic cavities filled with yellow-gray, pastry calcareous or chalky, milky material.
Microscopically, the active cellular phase is characterized by a central mass of amorphous or granular calcified material surrounded by a florid proliferation of histiocytes, multinucleated giant cells and variable fibrosis. Later, in the inactive phase, there is merely calcified material surrounded by dense fibrous tissue or a cystic space surrounded by calcium deposits. Occasional prominent small psammoma-like bodies or calcospherites are present.
Since calcification and ossification can be seen in many benign and malignant soft tissue lesions, the differential diagnosis for tumoral calcinosis is broad and includes all tumoral calcinosis – like lesions or processes that lead to abnormal dystrophic or metastatic calcium deposition in the soft tissues:
Acral tumoral calcinosis-like lesions consist of multiple cystic or cleft-like spaces bordered by histiocytes, osteoclast-like giant cells, with a variable inflammatory infiltrate and containing fibrin, granular calcific debris, and calcospherites. However, this lesion is small and is often located in tenosynovial/fascial tissue, with occasional dermal or intra-articular involvement. It can be associated with trauma, scleroderma, or long-standing osteoarthritis.
• Dystrophic calcification shows a similar microscopic picture as tumoral calcinosis, but usually is smaller and is associated with necrosis secondary to minor injury, ischemic necrosis or a necrotizing infectious process.
• Calcifying fibrous pseudotumor is a fibrous, slowly-growing and painless lesion that most often affects children and young adults. It is a generally well-circumscribed, non-encapsulated, hypocellular lesion composed of hyalinized fibrosclerotic tissue with a scattered lymphoplasmacytic infiltrate, sometimes forming lymphoid follicles as well as bland, sparse and dispersed spindle cells within the collagen. The characteristic feature is the presence of scattered psammomatous calcifications.
• Chondroma of soft parts may have similar features to tumoral calcinosis. However, chondroma usually involves the hands and feet and has a larger component of differentiated cartilage and lacks the amorphous material. The calcified material is granular, floccular or crystalline and often outlines the contours of the chondrocytes in a lace-like pattern.
• Calcifying aponeurotic fibroma is a rare, slow growing, small (usually less than 3 cm), painless tumor with fibroblasts palisading around chondroid or calcified nodules, usually in hands and feet of children or young adults. The calcifications are usually small and vary from fine granules or string-like deposits to large amorphous masses, often surrounded by radiating columns of chondrocyte-like cells.
• Calcifying tendinitis is a condition in which there are deposits of hydroxyapatite crystals in tendons, followed by calcification as well as macrophage and giant cell reaction. The lesion mostly involves tendons of rotator cuff especially that of supraspinatus.
• Calcifying synovial sarcoma. Calcification can be present to a varying degree in both monophasic and biphasic variants of synovial sarcoma. It may be inconspicuous and consist of a few small irregularly distributed spherical concretions, or it may be extensive and occupy a large portion of the neoplasm. The subset with extensive calcification occurs mainly in the lower extremity and is slowly-growing. Most synovial sarcomas show immunoreactivity for epithelial markers (CK and EMA), TLE1, CD99, BCL-2 and calponin. In addition, more than 90% of synovial sarcoma have a balanced reciprocal non-random translocation t(X;18)(p11;q11), with fusion of SSX and SS18 (SYT) genes.
In summary, tumoral calcinosis is a benign neoplasm characterized by abnormal peri-articular deposits of calcium hydroxyapatite around major joints. Although there are different familial and sporadic forms, the constellation of morphologic features is fairly consistent, which is characterized by a central mass of amorphous or granular calcified material surrounded by a florid proliferation of histiocytes, multinucleated giant cells and variable fibrosis.
Suggested Reading:
Hornick JL. Practical Soft Tissue Pathology: A Diagnostic Approach. Elsevier.
Laskin WB, Miettinen M, Fetsch JF. Calcareous lesions of the distal extremities resembling tumoral calcinosis (tumoral calcinosis-like lesions): clinicopathologic study of 43 cases emphasizing a pathogenesis-based approach to classification. Am J Surg Pathol 2007;31;15-25.
Carmichael KD, Bynum JA, Evans EB. Familial tumoral calcinosis: a forty-year follow-up on one family. J Bone Joint Surg Am 2009;91(3):664-71.
Olsen KM, Chew FS. Tumoral calcinosis: pearls, polemics, and alternative possibilities. Radiographics 2006;26(3):871-85.
Goldblum JR, Folpe AL and Weiss SW. Enzinger & Weiss’s Soft Tissue Tumors: 6th ed. 2014, Elsevier.