History: A 5-year-old girl presented with cardiac conduction problem. Workup revealed a mass in the right atrium.
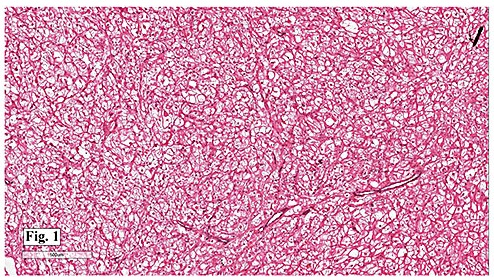
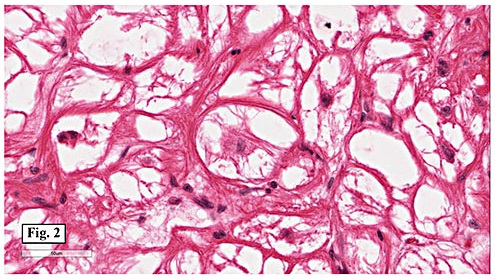
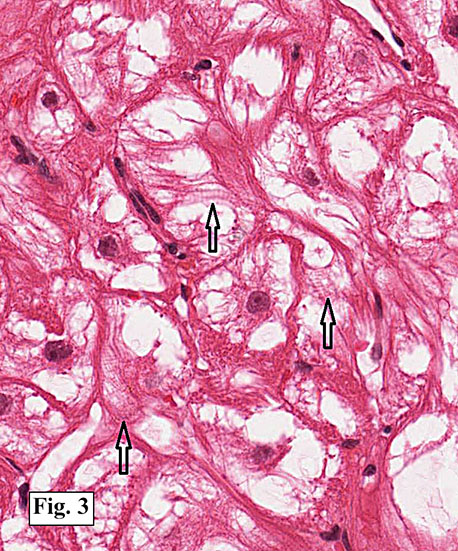
Microscopically, the mass consisted of enlarged myocytes with abundant, vacuolated or clear cytoplasm (Fig. 1). At higher magnification, the tumor cells had strands of cytoplasmic myofibrils radiating from the central nucleus to the cell periphery forming so-called “spider cells†(Fig. 2). Cross striations were commonly seen (Fig. 3). Neither necrosis nor mitotic figures were present.
{kind=link}
{kind=link}
{kind=link}
Diagnosis: Rhabdomyoma of the atrium
Li Lei, M.D. and Donald R. Chase, M.D.
Department of Pathology and Human Anatomy
Loma Linda University Medical Center, Loma Linda, California
California Tumor Tissue Registry, Loma Linda, California
Discussion: Primary cardiac tumors are rare. In the young pediatric group, the commonest cardiac tumors are rhabdomyomas (45-63%), followed by fibromas (20%) and myxomas (10-15%); while in teenagers and adults, myxomas account for about two-thirds of primary cardiac tumors.
As its name implies, rhabdomyoma is a benign tumor of striated muscle differentiation. It is considered a hamartoma or malformation other than a true neoplasm, largely because the tumor cells have a very low proliferative index … essentially, they do not proliferate.
Cardiac rhabdomyoma almost exclusively affects infants and young children. About 75% of cases occur before the age of one year. It can occasionally be seen as early as 20 weeks gestational age. Arrhythmia is the most common presentation, though many patients are asymptomatic. Lethal arrhythmia or valvular orifice obstruction can cause sudden cardiac death.
Approximately 51-88% of cardiac rhabdomyomas are associated with tuberous sclerosis, and it may be the earliest sign of tuberous sclerosis in a family. On the other hand, about 43-72% of children with tuberous sclerosis have or will develop cardiac rhabdomyomas. A minority of cases are sporadic. Occasionally, association with folliculin mutation and Birt Hogg Dube syndrome has been reported.
Cardiac rhabdomyomas are often multiple, particularly those associated with tuberous sclerosis. Up to 10% of cases are solitary. It typically presents as intramural lesion in the ventricles, but may protrude into the ventricular cavity as a pedunculated or sessile mass. Interventricular septum and atria can also be involved. Macroscopically, it is well-circumscribed but nonencapsulated, with a homogeneous whitish or grayish cut surface.
Histologically, the cells are big and polygonal. Large cytoplasmic vacuoles are separated by strands of cytoplasm extending radially between nucleus and cell membrane, giving a characteristic “spider cell†appearance. The vacuoles are rich in glycogen which is PAS positive and diastase sensitive. Cross-striations are also routinely seen. Mitotic figures and necrosis are absent.
Ultrastructurally, the cells have abundant glycogen and rare, small mitochondria. Cellular junctions resembling intercalated disks are all around the periphery instead of exclusively at the cell poles as seen in normal cardiomyocytes.
Immunohistochemically, the tumor cells express muscle specific actin (MSA), myoglobin, desmin and vimentin. As with other tumors associated with tuberous sclerosis, cardiac rhabdomyoma is often positive for HMB45 but is usually negative for S100.
The differential diagnosis of cardiac rhabdomyoma includes:
• Histiocytoid cardiomyopathy is extremely rare in patients over the age of 2 years. It presents as subendocardial yellowish nodules or plaques. The large polygonal cells have coarsely granular, foamy to pale eosinophilic cytoplasm resembling histiocytes. They show cholinesterase immunoreactivity but do not express histiocytic antigen(s). The granules are bizarre mitochondria under electron microscope.
• Cardiac hibernoma consists of granular to multivacuolated adipocytes, which are typically S100 positive. The cytoplasmic vacuoles are small and stain for neutral fat. “Spider cells†and cross-striations are lacking.
• Cardiac myxoma often occurs on the left side of the atrial septum. It arises from endocardium as a polypoid mass and does not involve underlying myocardium. Cut surface is gelatinous. Perivascular cuffing by tumor cells within myxoid stroma is commonly seen.
• Cardiac rhabdomyosarcoma is anecdotal in children, with only a few cases reported in the literature. Invasive biologic behavior and malignant histologic features help differentiate. Extensive clinical workup excluding metastasis is necessary before rendering the diagnosis.
Cardiac rhabdomyoma has a tendency to regress spontaneously over time, especially throughout the first year. Approximately 54-100% of cases undergo complete or partial regression. Surgery is therefore reserved for seriously symptomatic patients. If the risk of radical resection outweighs the benefit, a partial resection may be satisfactory given the regressing potential of cardiac rhabdomyoma. In inoperable cases, mammalian target of rapamycin (mTOR) inhibitors such as everolimus and sirolimus have been tried and appear promising.
Suggested Reading:
Goldblum J, Folpe A, Weiss S. Enzinger & Weiss’ Soft Tissue Tumors, 6th ed: Philadelphia, Elsevier Inc, 2014; 591-2.
Suvarna SK. Cardiac Pathology: A Guide to Current Practice. Springer 2013; 201-21.
Basso C, Valente M, Thiene G. Cardiac tumor pathology. Springer 2013; 59-62.
Burke A, Virmani R. Pediatric heart tumors. Cardiovasc Pathol. 2008;17:193-8.
Bondavalli D, White SM, Steer A, Pflaumer A, Winship I. Is cardiac rhabdomyoma a feature of Birt Hogg Dubé syndrome? Am J Med Genet A. 2015;167A:802-4.