History: A 17 year-old male presented with significant hepatosplenomegaly and a mass in the right lobe of the liver. He had been born with a hypoplastic left heart and diagnosed with velocardiofacial syndrome. At 13 weeks of age he received a heart transplant and was placed on immunosuppressive therapy including; rapamune (sirolimus) and prograf (tacrolimus). Eleven years later he presented with fever, abdominal distension and respiratory distress. A CT scan of his chest was performed and revealed multiple enlarged lymph nodes in the right neck. A biopsy was performed.
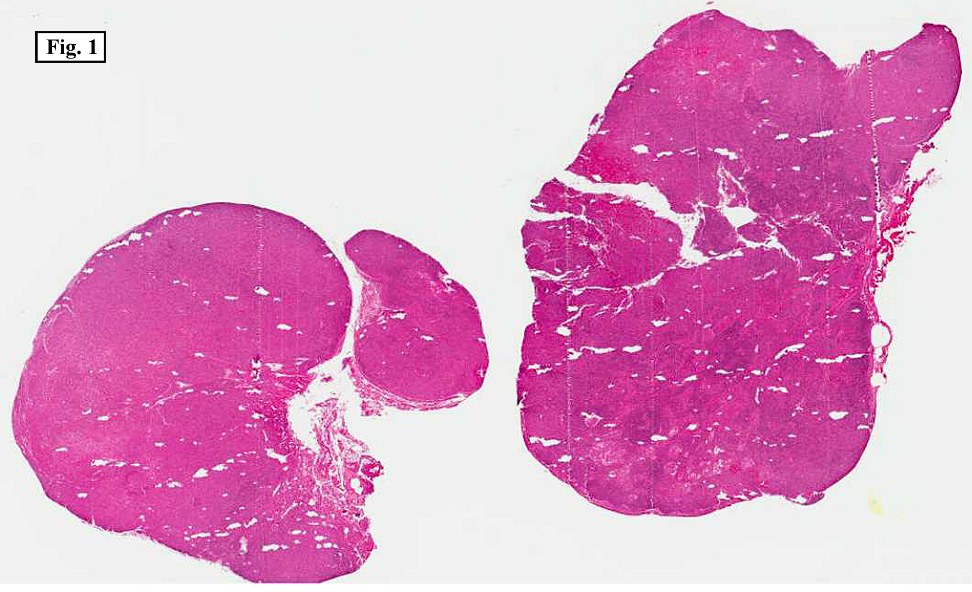
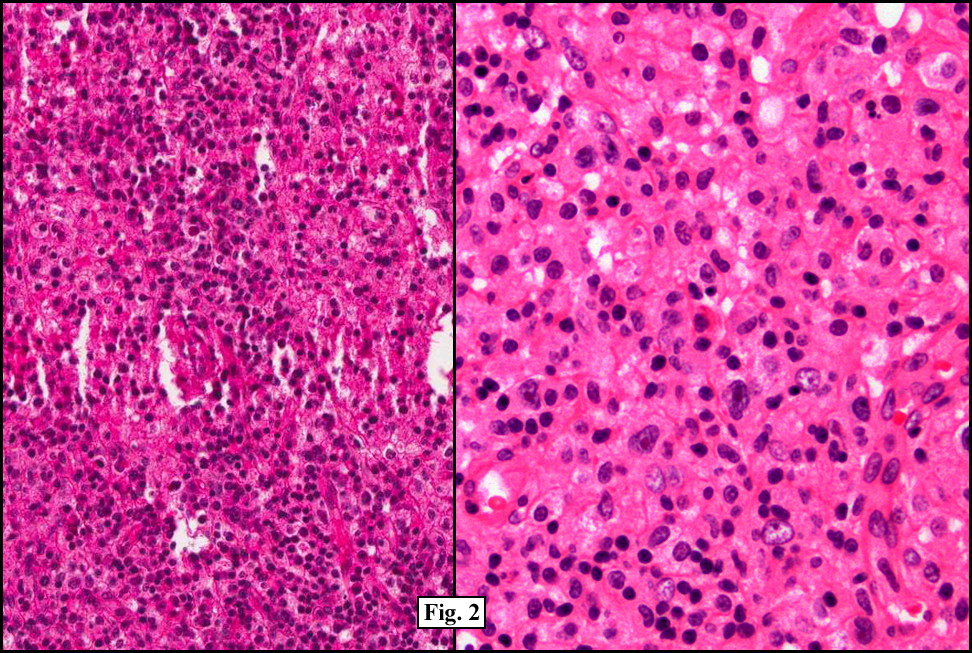
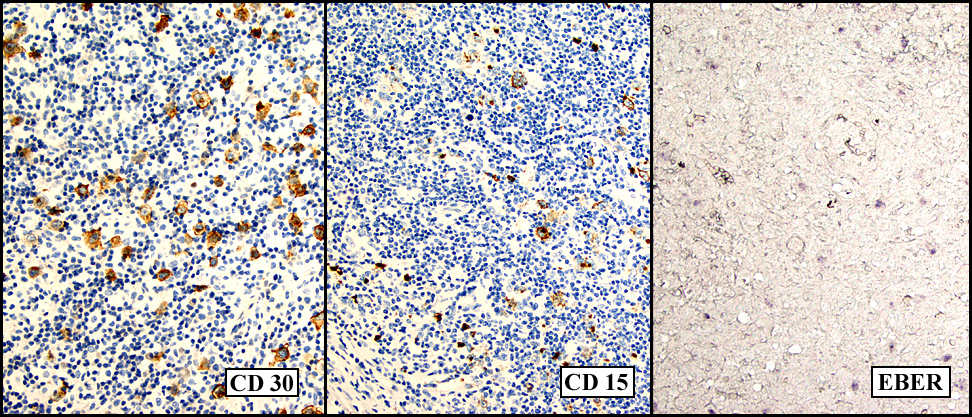
Microscopically, there was complete effacement of normal lymph node architecture (Fig. 1) by a heterogeneous population of large, pleomorphic cells with abundant basophilic cytoplasm, one to multiple nuclei, and prominent eosinophilic nucleolus consistent with Hodgkin Reed-Sternberg (HRS) cells, in a background of small lymphocytes, rare histiocytes, plasma cells and eosinophils (Fig. 2). Immunohistochemically, the tumor cells were positive for CD 30, CD 15, EBER (Fig. 3) and weakly positive for PAX-5, and negative for CD 3, CD 20, CD 43 and CD 45. Flow cytometric studies showed no aberrant antigen expression.
{kind=link}
{kind=link}
{kind=link}
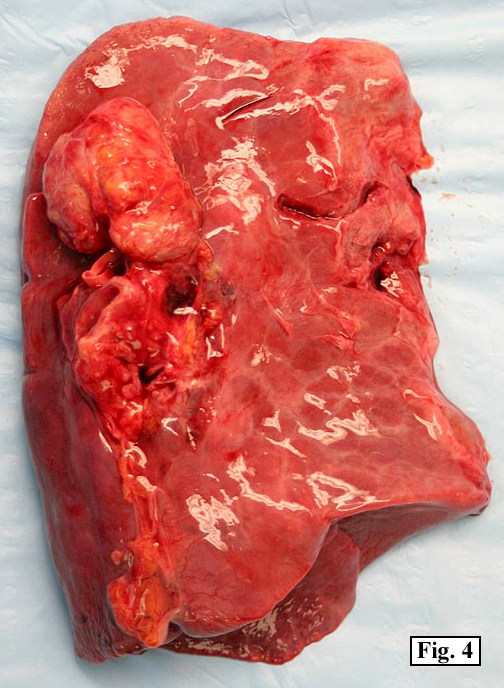
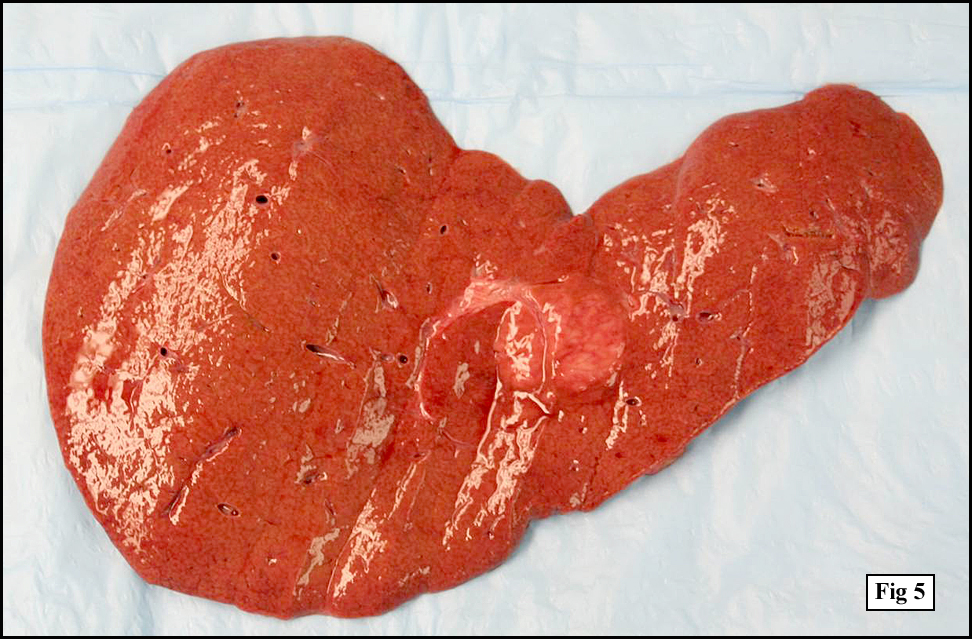
The patient’s immunosuppressive therapy was reduced and his lymphadenopathy resolved but he subsequently presented with signs of acute renal failure. A chest and abdominal CT scan revealed a mass in the right lobe of liver. Over the course of his hospitalization he developed sepsis, respiratory failure and eventually expired. On autopsy, prominent mediastinal (hilar and paratracheal) lymph nodes with a fleshy-tan cut surface were identified (Fig. 4). Also, a distinct 3.5 x 3.0 cm fleshy tan nodule was present within the mid-right lobe of the liver (Fig. 5).
{kind=link}
{kind=link}
Microscopically, sections of the lymph nodes and the liver mass were identical to the prior lymph node biopsy. There was complete effacement of normal architecture by lymphohistiocytic infiltrate consisting of large HRS cells in a background of small lymphocytes, occasional histiocytes, plasma cells and eosinophils. A limited panel of immunostains was performed which revealed the tumor cells to be positive for CD30, CD15 and negative for CD20.
Diagnosis: “Recurrent Classical Hodgkin lymphoma type Post-transplant lymphoproliferative disorderâ€
Hannah H. Wong, MD, Jun Wang, MD, Donald R. Chase, MD.
Department of Pathology, Loma Linda University Medical Center,
Loma Linda, California
California Tumor Tissue Registry, Loma Linda, California
Discussion: Classical Hodgkin lymphoma type Post-transplant lymphoproliferative disorder (cHL-PTLD) is a rare subtype of PTLD, first described in 1974 in a kidney transplant patient. Post-transplant lymphoproliferative disorders (PTLD) are a group of lymphoid or plasmacytic lesions which develops in roughly 1-5 % of transplant patients secondary to immunosuppression following solid organ, bone marrow and/or stem cell transplantation.
PTLDs are classified by the 2008 WHO classification of Tumors of Haematopoietic and Lymphoid tissues into four main categories:
(1) Early lesions (plasmacytic hyperplasia, infectious mononucleosis-like lesion)
(2) Polymorphic PTLD
(3) Monomorphic PTLD (including B-cell and T-cell neoplasms)
(4) Classical Hodgkin lymphoma-type PTLD
cHL-PTLD has a clinical behavior and morphological presentation identical to classical Hodgkin lymphoma (cHL) and was originally classified in the same category as Hodgkin lymphoma-like PTLD. The 2008 WHO; however, redefined this fourth category of PTLD as classical Hodgkin lymphoma type PTLD and the “Hodgkin lymphoma-like PTLD†is no longer included in this category but rather classified as Polymorphic or Monomorphic B-cell PTLD
Epstein-Barr virus (EBV), a human herpes virus, has been implicated in the development of the majority of PTLD cases, from 60% to up to 100% positivity in early lesions. In fact, only 23-42% of PTLD cases have been found to be EBV negative. EBV infects human lymphocytes, induces cellular proliferation and inhibits apoptosis resulting in the development of malignancies. This virus can remain dormant within cells without viral replication for an in-determined period of time. B-lymphocytes have an internal defense mechanism which prevents viral replication, but allows the expression of a restricted set of viral genes called “latency†genes. These latency genes code for six nuclear and three membrane associated proteins which interfere with cellular control, proliferation and differentiation of B-lymphocytes.
Four latency programs have been described based on the expression of different viral gene combinations in correspondence to the different stages of B-cell activation and differentiation. Each latency program has been associated with a different group of malignancies, except for Latency 0. Latency I is associated with Burkitt lymphoma, latency II with cHL, peripheral T-cell lymphoma, nasal T/NK cell lymphoma, nasopharyngeal carcinoma, and lymphoepithelioma (stomach, thymus). Latency III is associated with infectious mononucleosis, AIDS-related immunoblastic B-cell lymphoma, and PTLD. Table I gives a summary of the four EBV latency programs, viral gene expression and associated diseases.
Table I. EBV latency programs, expressed viral genes and associated diseases
| Latency program | Expressed viral genes | Disease |
| Latency 0 | None | None |
| Latency I | LMP-2A/EBNA-1 | Burkitt lymphoma |
| Latency II | EBNA-1, LMP-1, LMP-2A, -2B | cHL, peripheral T-cell lymphoma, nasal T/NK cell lymphoma |
| Latency III | EBNA-1, -2, -3, -4, -5, -6, LMP-1, -2A, -2B | Infectious mononucleosis, AIDS-related immunoblastic B-cell lymphoma, PTLD |
Patients with cHL-PTLD present with signs and symptoms identical to classical Hodgkin Lymphoma (cHL) such as fever, weight loss, night sweats and lymphadenopathy. It involves nodal or extranodal sites with a predilection for male and pediatric/adolescent patients. cHL-PTLD has been found to develop much later, even years later in comparison to the other forms of PTLD and has been found more commonly in patients with renal transplants.
Morphologically, cHL-PTLD is virtually identical to cHL. The involved tissue shows a mixed population of small to medium-sized lymphoid cells admixed with scattered large mononucleated, binucleated, and/or multinucleated Hodgkin Reed-Sternberg (HRS) cells, as well as occasional histiocytes, plasma cells, and rare eosinophils in the background. Immunohistochemically, the HRS-cells show the classic expression of CD30 and CD15 antigens, but do not express CD45, CD20 and CD79a. PAX-5 may be positive in some HRS cells, but does not change the diagnosis. EBV-encoded RNA (EBER) in situ hybridization is positive only in the RS-cells. The HRS-cells will also reveal expression of Type II EBV latency.
The main and most important differential diagnosis of cHL-PTLD is Hodgkin lymphoma-like PTLD (HL-like PTLD). The distinction between these two entities is of importance since the biology and treatment of these two entities are quite different. HL-like PTLD has been found to be more similar in its clinical behavior to monomorphic and polymorphic B-cell forms of PTLD than to cHL. Clinically, cHL-PTLD and HL-like PTLD may present with similar symptoms of lymphadenopathy, fever, night sweats and splenomegaly, generally following years of immunosuppression. The morphological features of HL-like PTLD are difficult to differentiate from cHL-PTLD as it also shows large atypical HRS-like cells with a background of small lymphoid cells, occasional histiocytes, plasma cells, and eosinophils. Immunohistochemically, the staining patterns between the two are more distinctive. The HRS-like cells in HL-like PTLD are positive for CD30 and CD20 as well as CD45, but generally negative for CD15. This is in contrast to cHL-PTLD where the HRS-like cells are CD30 and CD15 positive but CD20 and CD45 negative. The pattern of EBER positivity also differs in these two entities, with EBER positivity in the large atypical HRS-like cells, as well as, surrounding by-stander small lymphocytes in HL-like PTLD but only positive in the HRS-cells in cHL-PTLD. HL-like PTLD has also been shown to express a Type III EBV latency pattern, similar to the polymorphic and monomorphic B-cell types of PTLD. HL-like PTLD has been found to respond well to treatment regimens for monomorphic B-cell PTLD such as reduction of immunosuppresion, along with anti-CD20-antibody (rituximab and combinations of chemoimmunotherapy for non-Hodgkin lymphoma (NHL).
Despite the re-classification and better understanding of cHL-PTLD as a distinct type of PTLD with features identical to cHL, there is currently no established treatment protocol. In the reported cases of cHL-PTLD, reduction/withdrawal of immunosuppression along with chemotherapy and radiotherapy have been utilized. Patients treated with standard cHL chemotherapy (such as doxorubicin, bleomycin, vinblastine and dacarbazine [ABVD]) and radiotherapy have been found to do better than those treated as monomorphic PTLD. It has been suggested that cHL-PTLD may respond best to standard cHL treatment protocols.
Table II. Comparison of cHL-PTLD and HL-PTLD
| cHL-PTLD | HL-like PTLD | |
| Clinical Features | Fever, lethargy, lymphadenopathy | Fever, lethargy, lymphadenopathy |
| Histology | HRS cells in a background of small lymphocytes, occasional histiocytes, plasma cells and eosinophils | HRS-like cells in a background of small lymphocytes, occasional histiocytes, plasma cells and eosinophils |
| Immunohistochemistry | CD30+, CD15+, CD45-, CD20-/+, CD79a- | HRS-like cells: CD30+, CD15-, CD45+, CD20+/-, CD79a+ |
| EBV status | EBER+ in HRS-cells only | EBER+ variably positive in HRS-like cells AND by-stander small lymphocytes |
| EBV latency | Type II latency pattern | Type III latency pattern |
| Treatment | ABVD* or other chemotherapies used for Hodgkins lymphoma | Reduced immunosuppression, anti-CD20-antibody (rituximab), non-Hodgkin lymphoma chemotherapies (e.g CHOP** |
* ABVD – doxorubicin, bleomycin, vinblastine, dacarbazine
**CHOP – cyclophosphamide, adriamycin, vincristine, prednisone
Suggested reading:
Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele N, Vardiman JW, Eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: IARC; 2008.
Pitman SD, Huang Q, Zuppan CW, Rowsell EH, Cao JD, Berdeja JG, Weiss LM, Wang J. Hodgkin lymphoma-like posttransplant lymphoproliferative disorder (HL-like PTLD) simulates monomorphic B-cell PTLD both clinically and pathologically. Am J Surg Path.2006;30:470-476