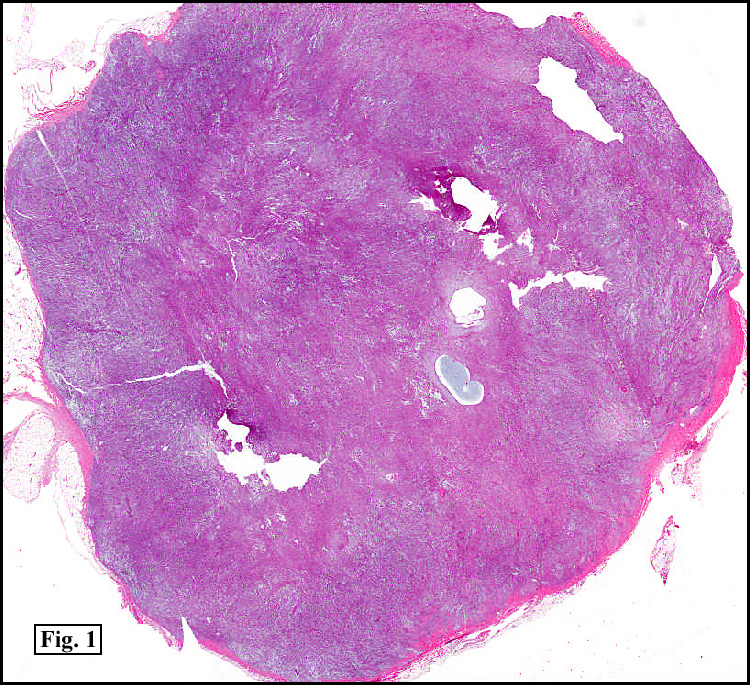
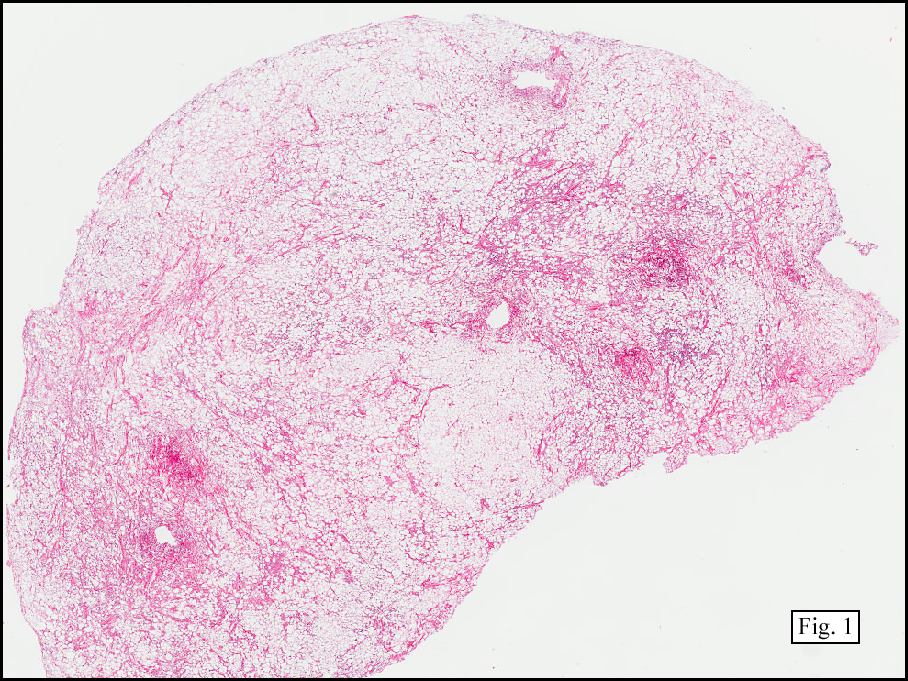
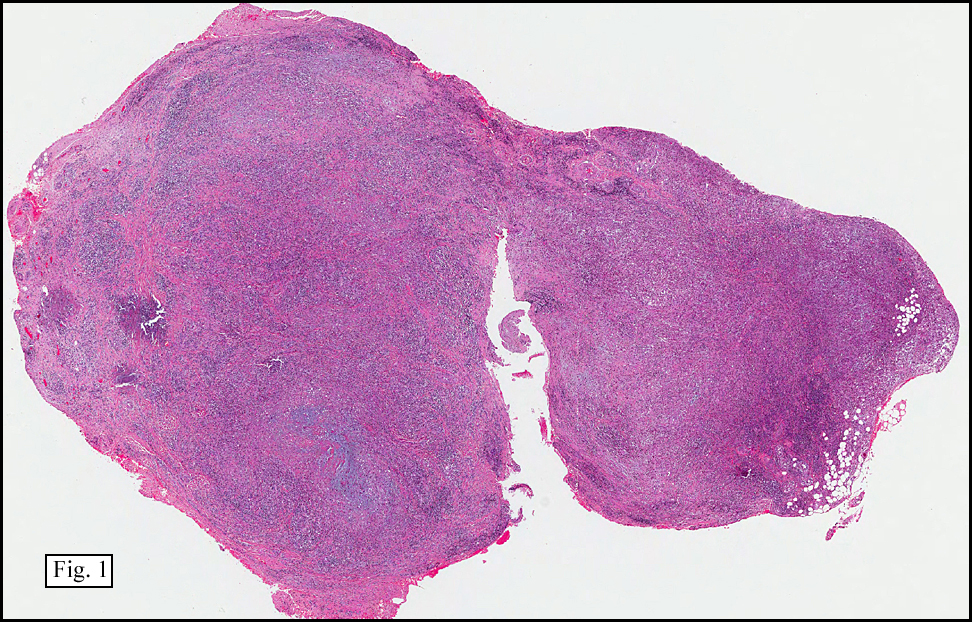
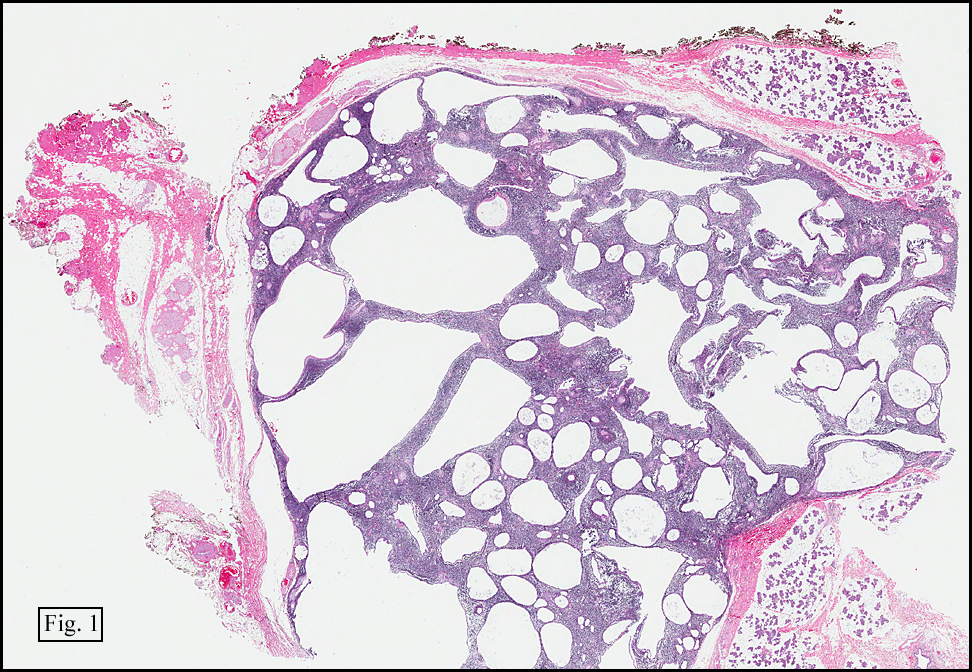
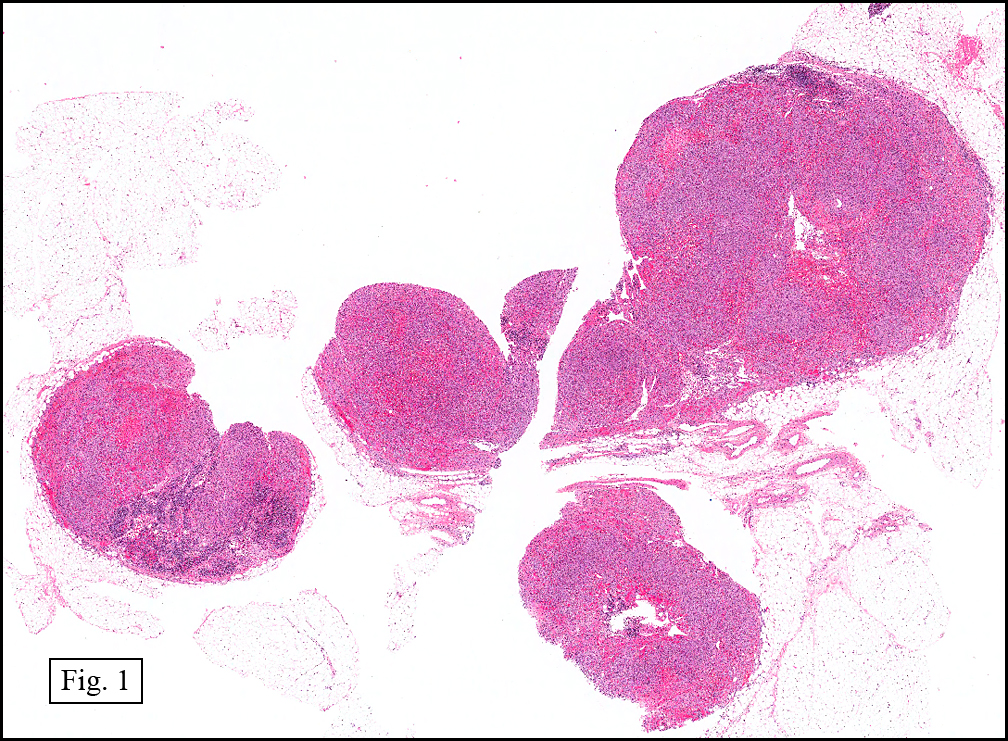
History: A 16-year-old young woman presented with a six month history of a posterior left thigh nodule. The mass was associated with mild pain and swelling. Surgery revealed a 3 cm well-defined and partially encapsulated nodule without apparent involvement of adjacent muscle (Fig. 1).
{kind=link}
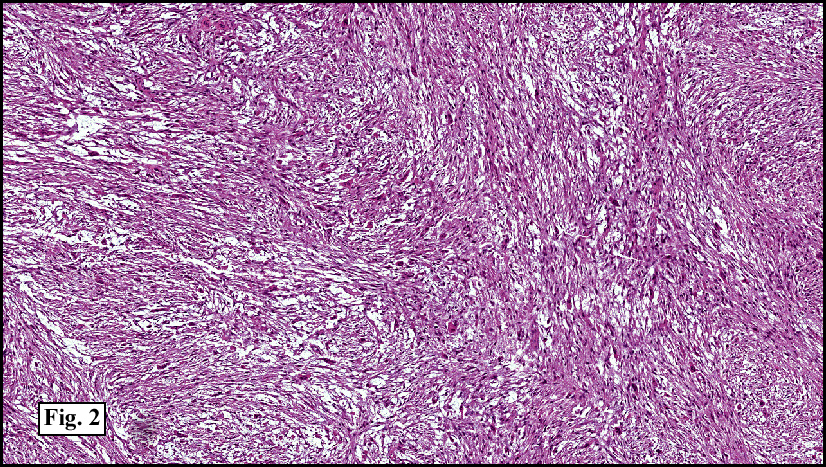
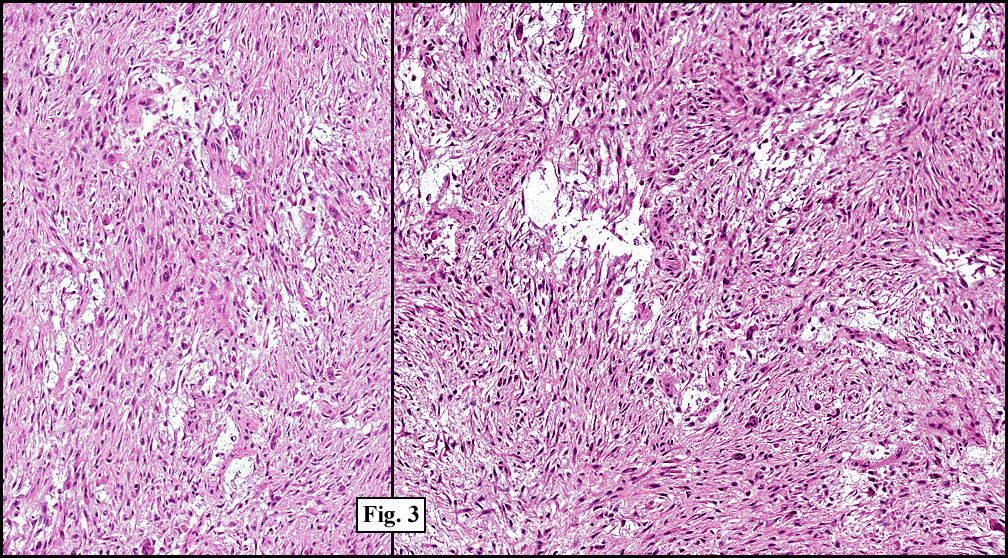
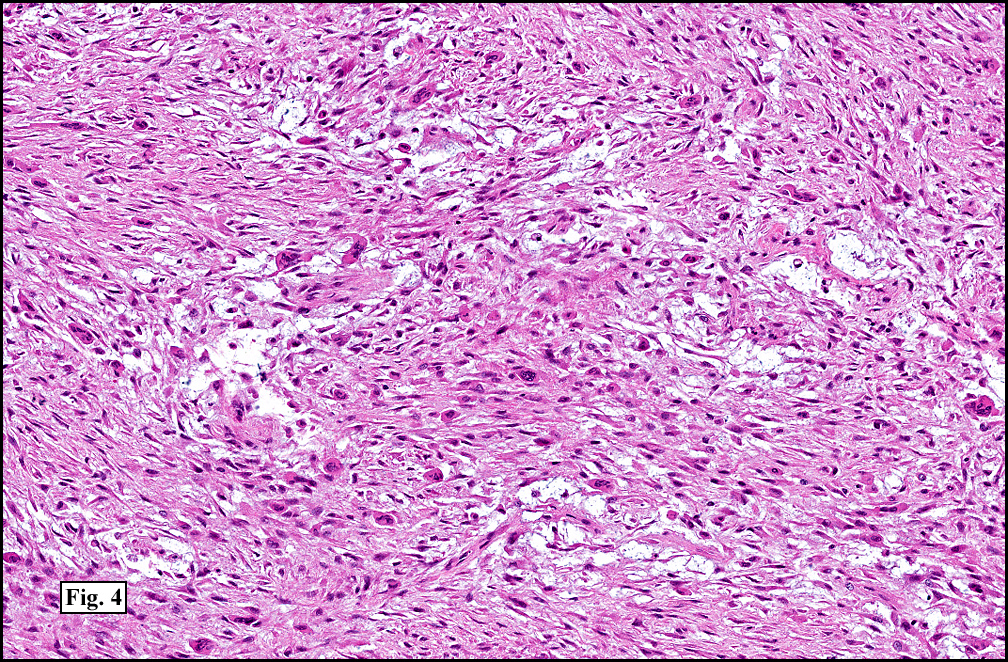
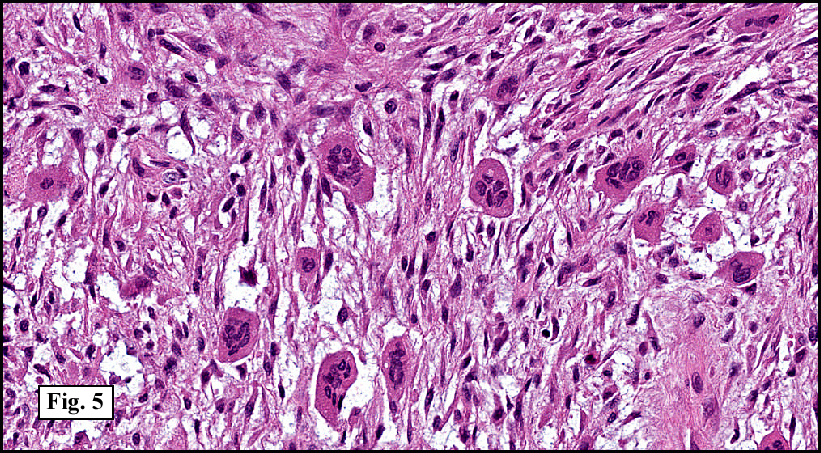
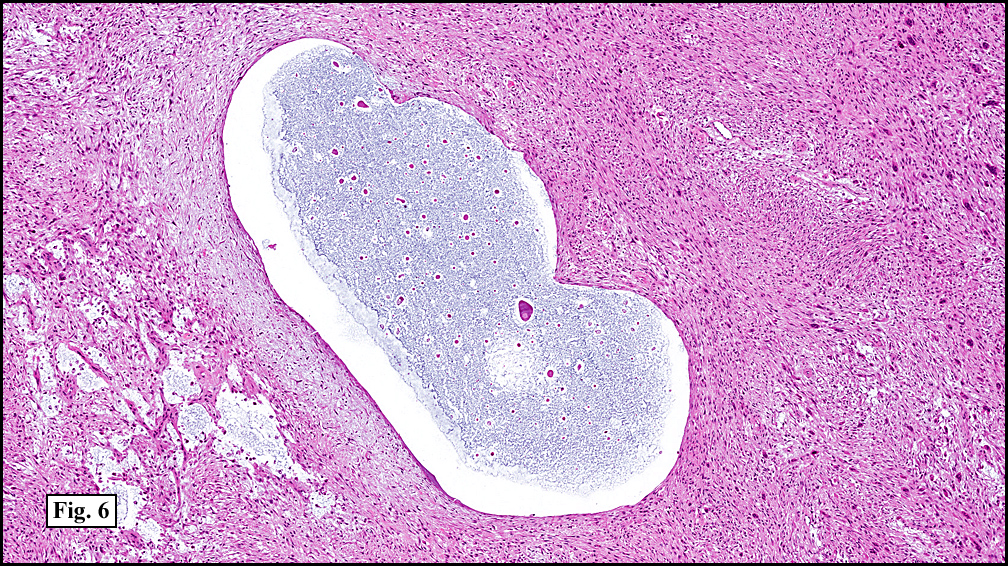
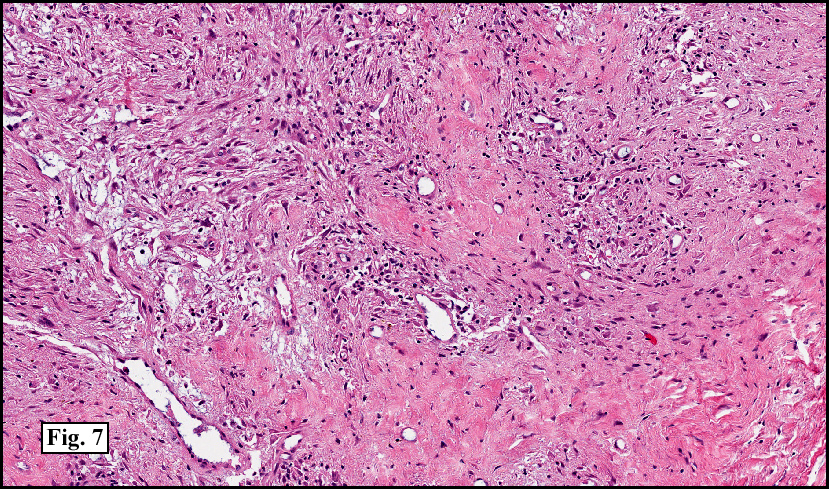
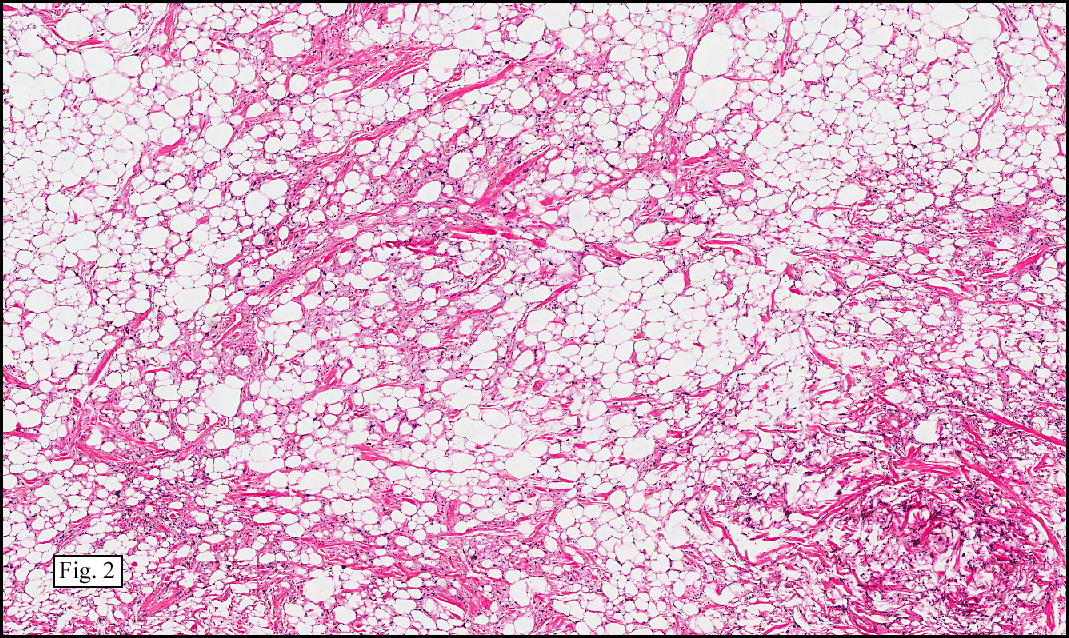
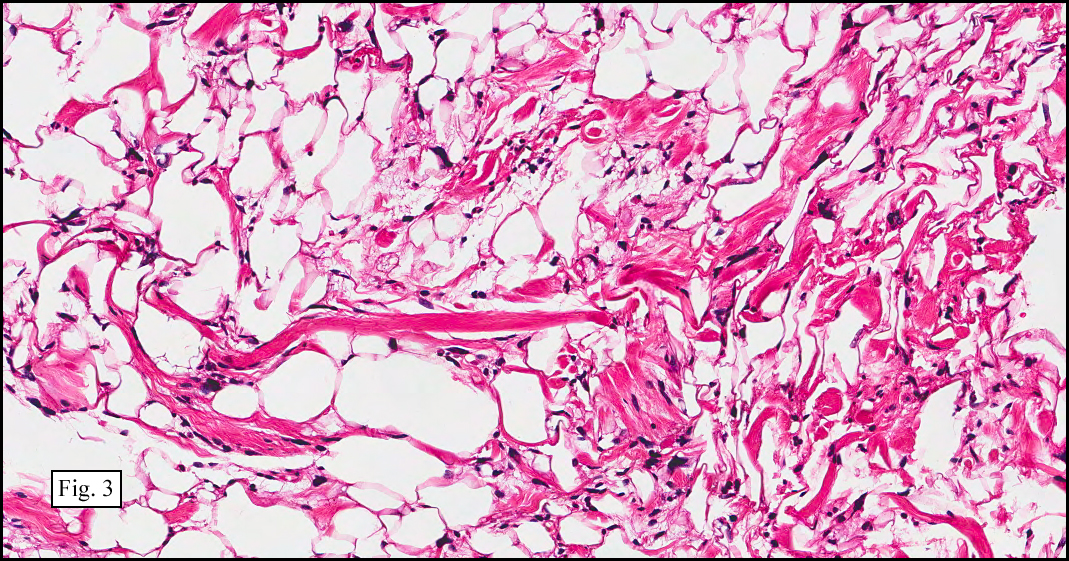
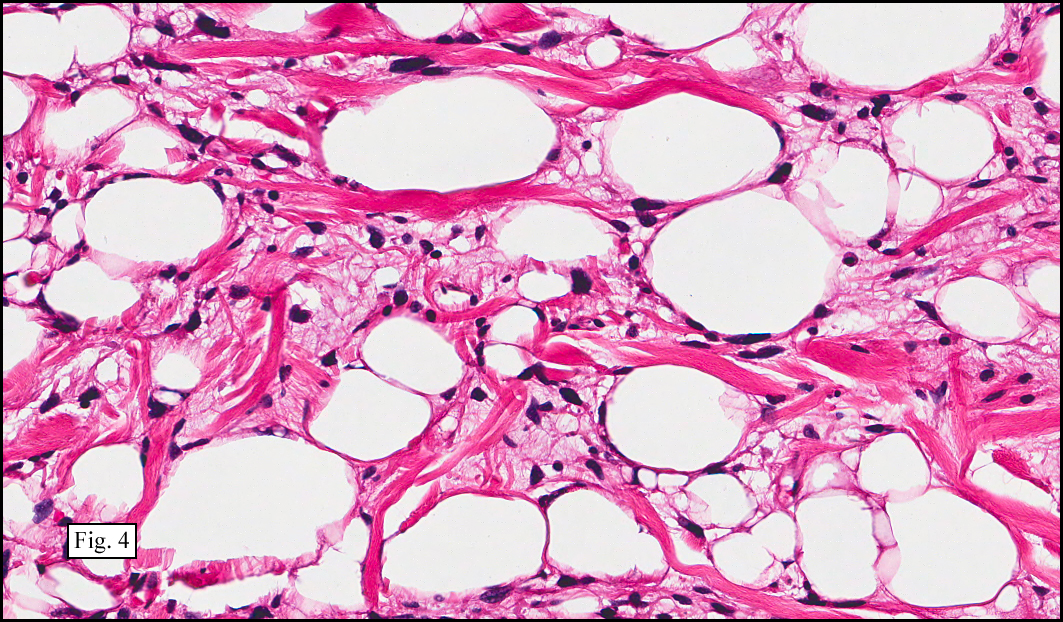
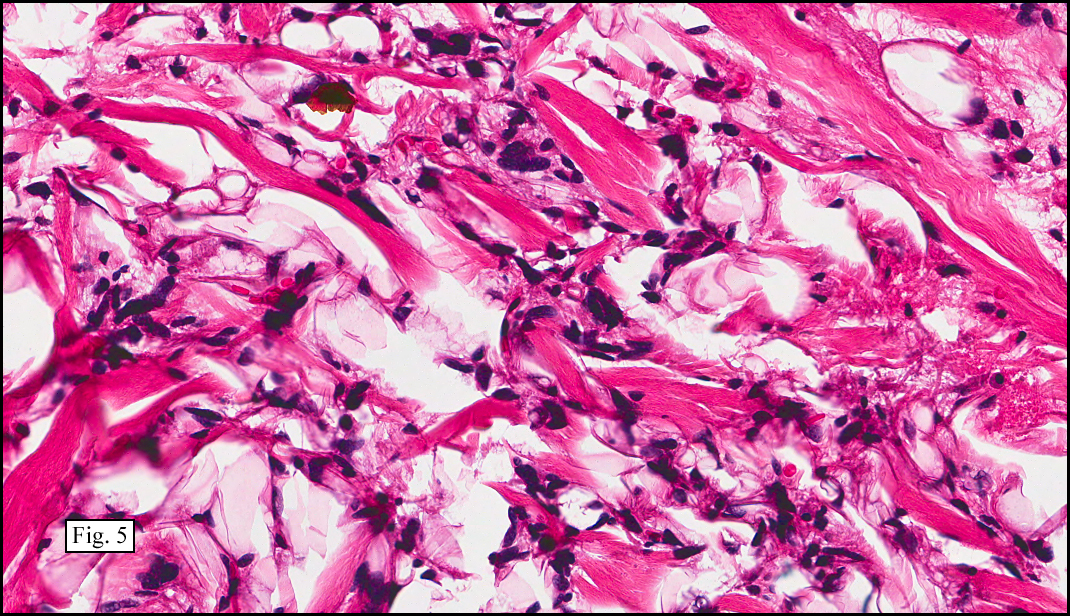
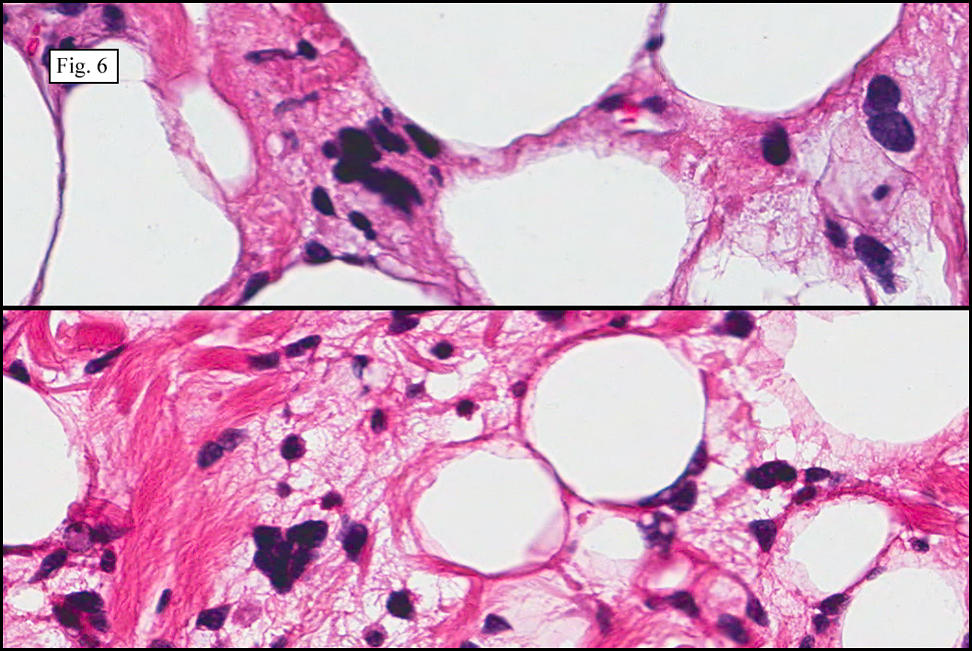
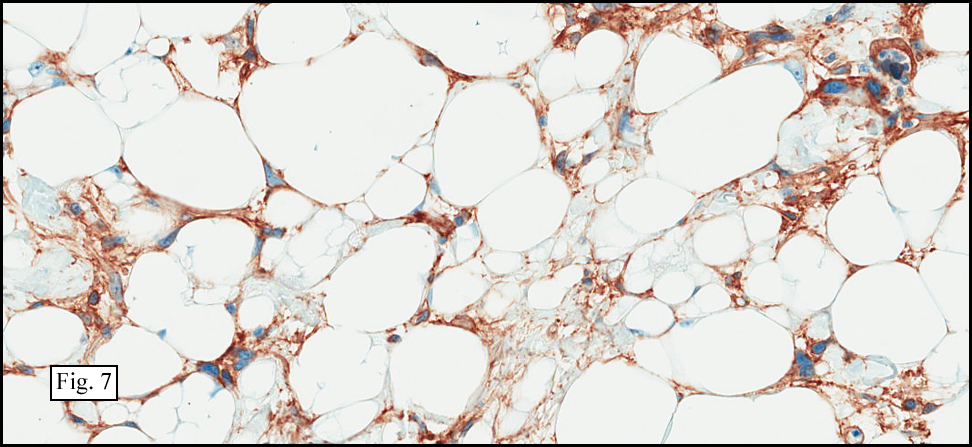
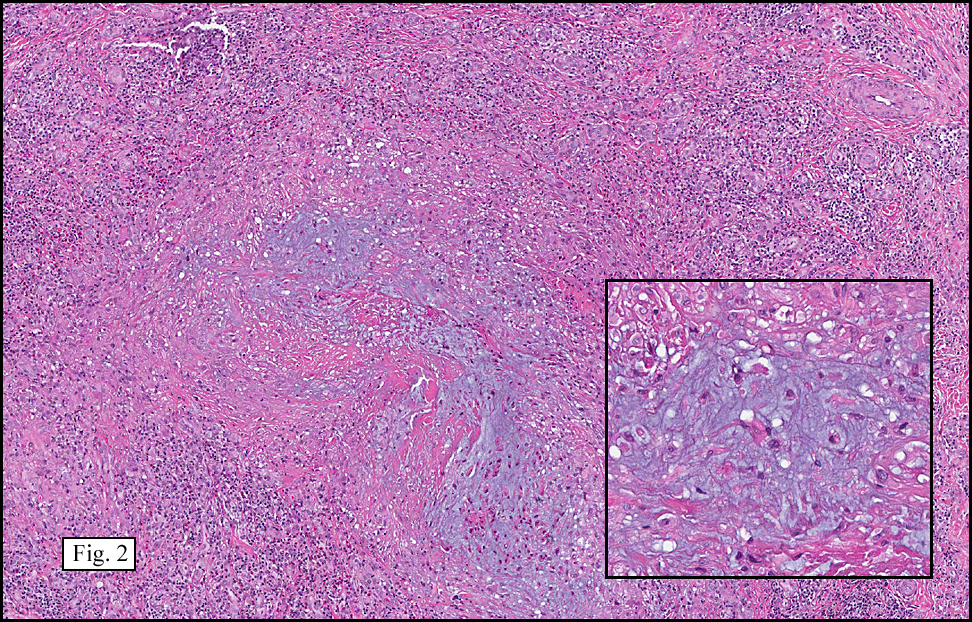
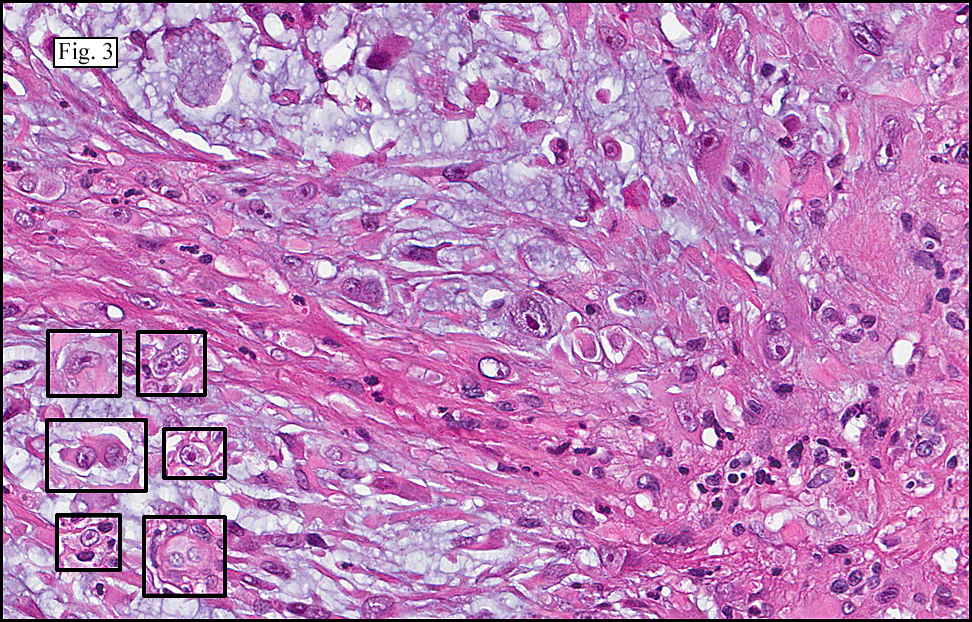
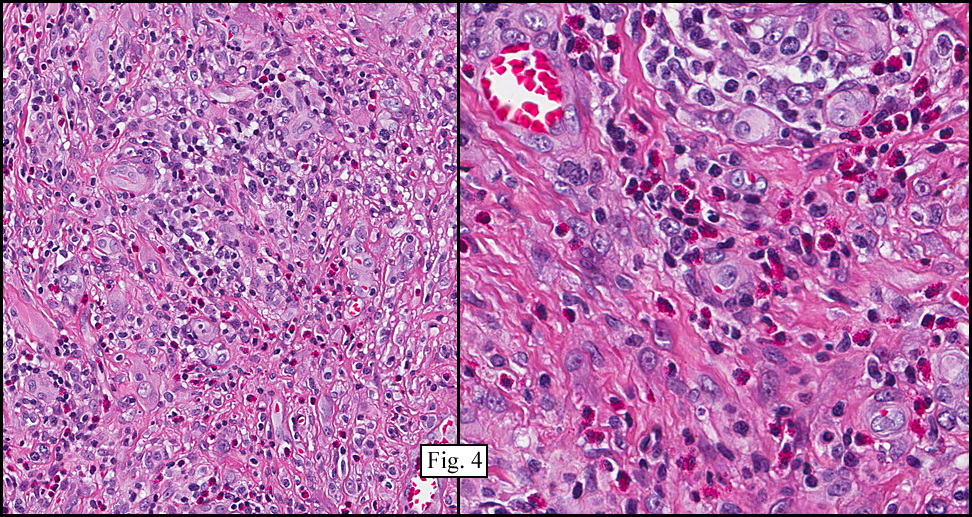
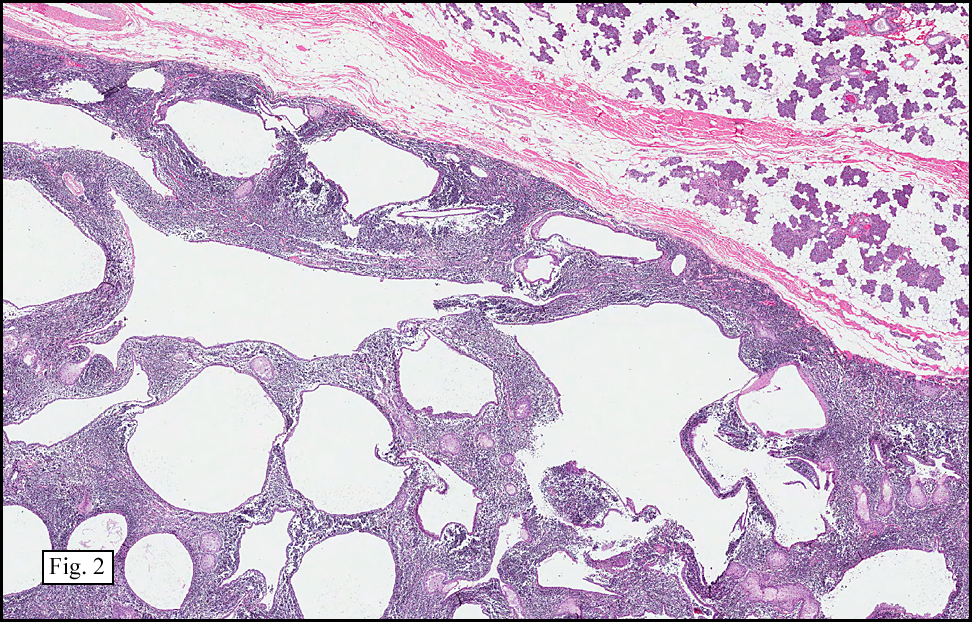
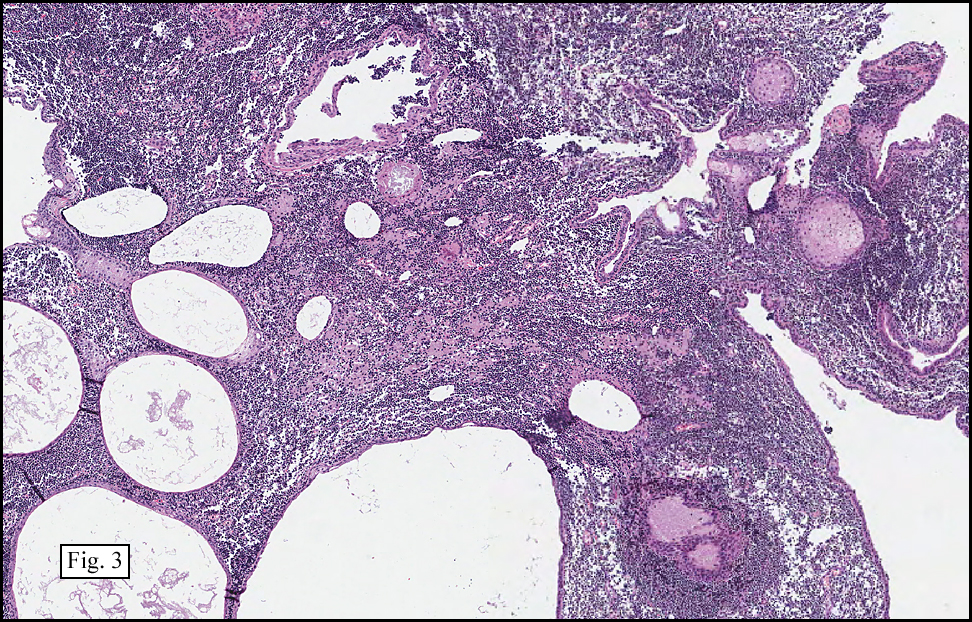
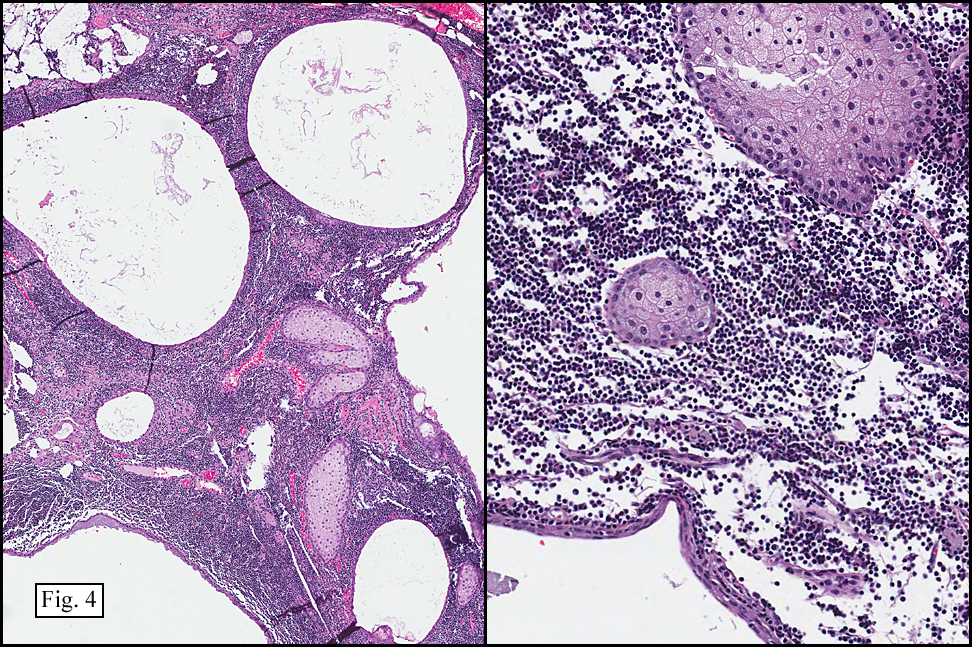
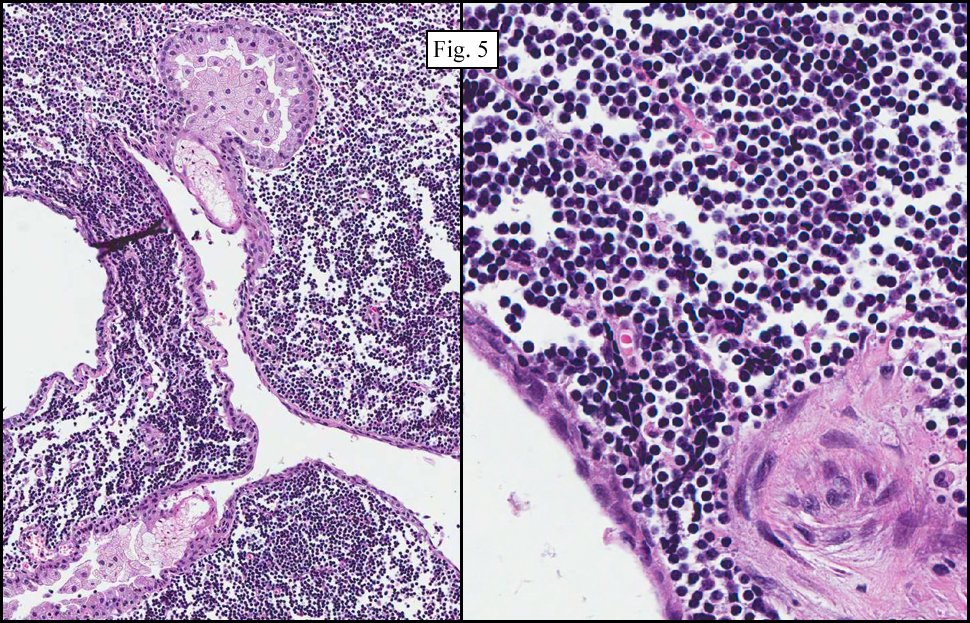
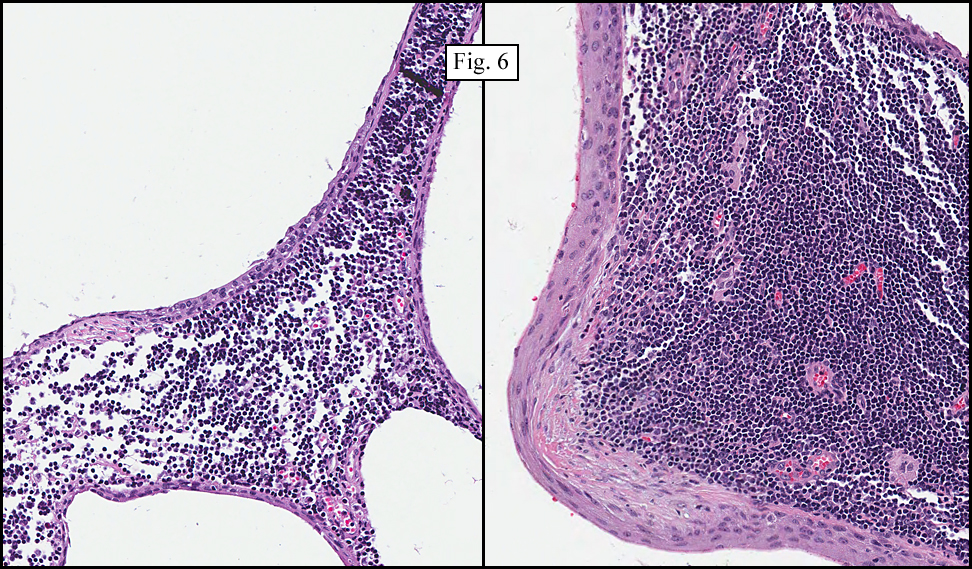
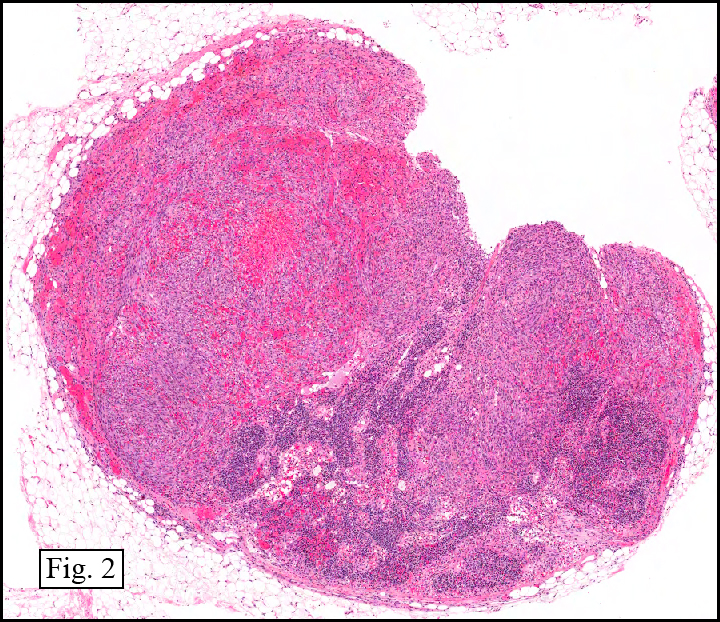
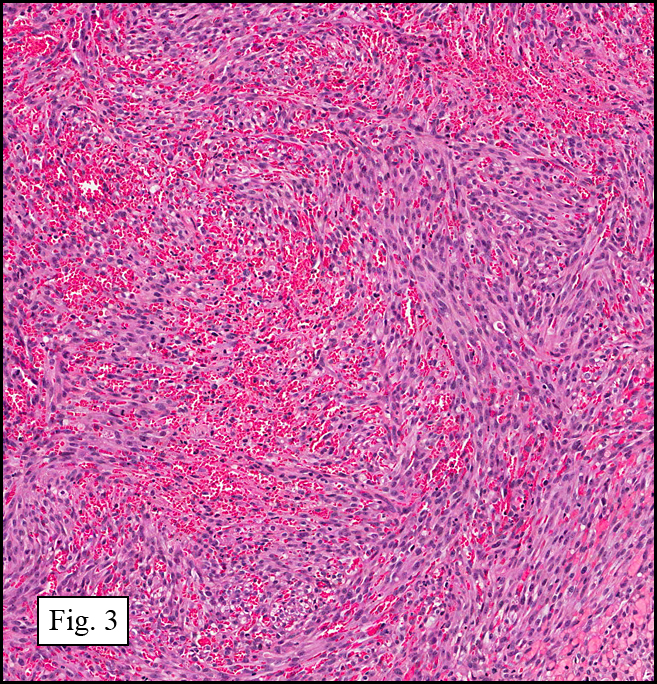
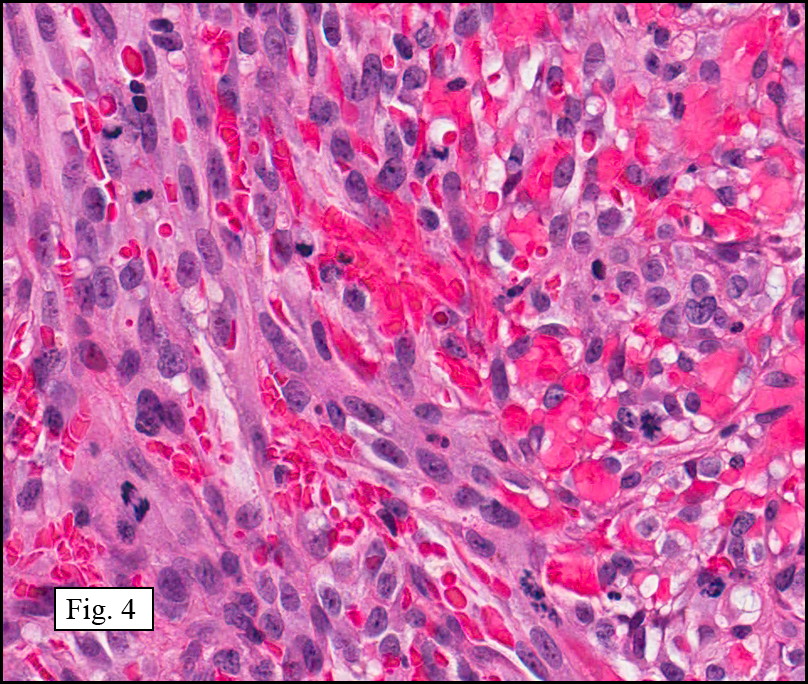
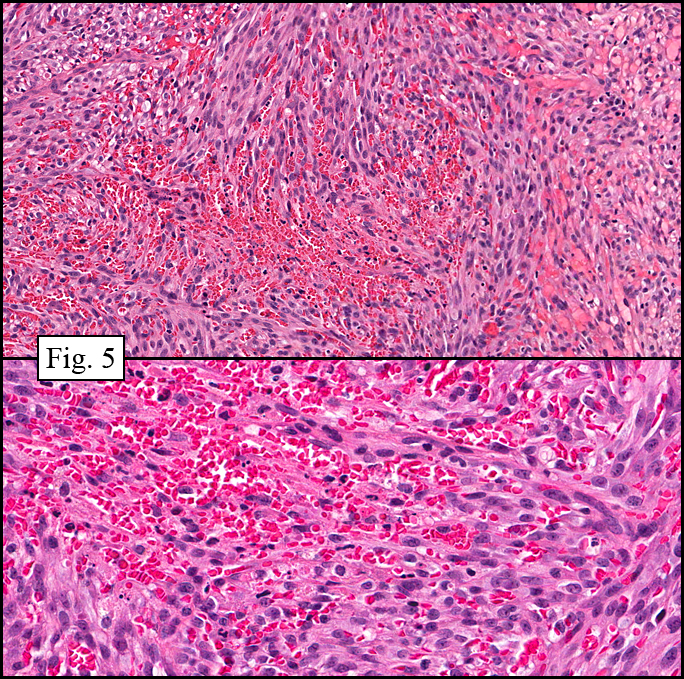
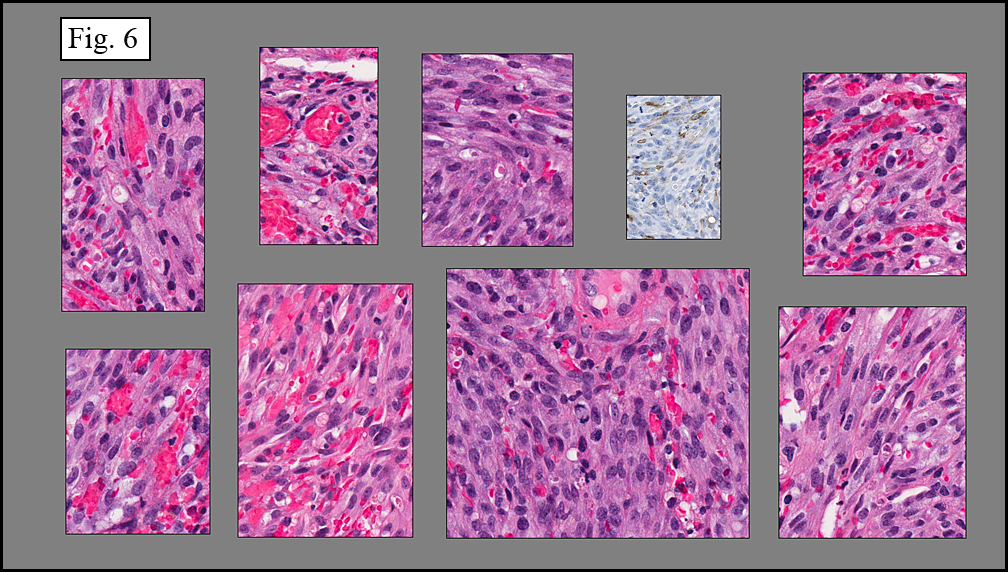
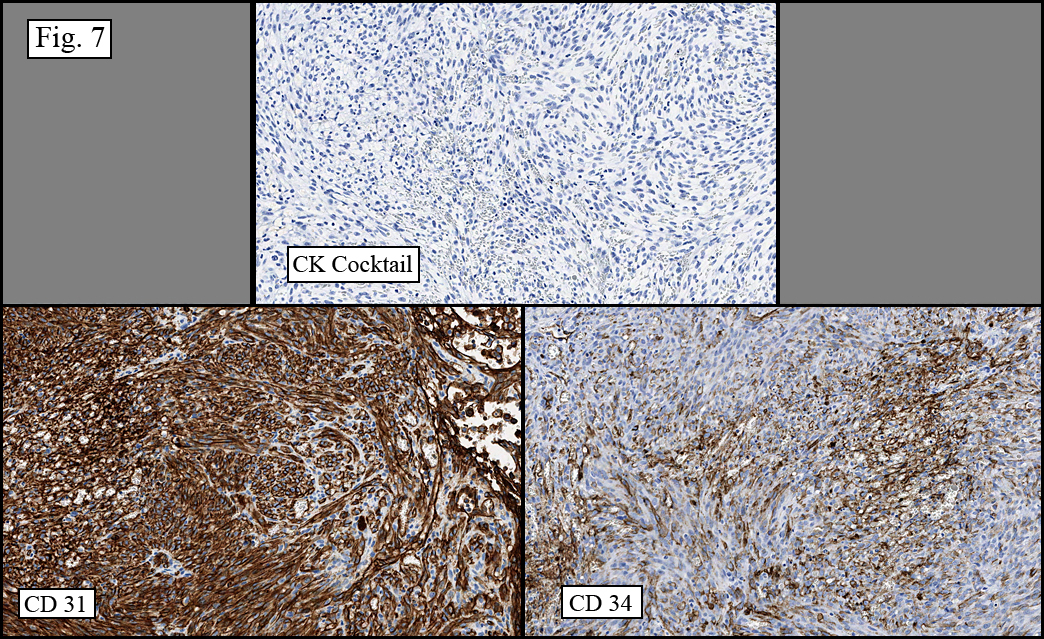
Microscopy showed spindled to stellate-shaped cells with a feathery or fibrillary appearance in a myxoid background (Fig. 2, 3, 4). Scattered giant cells were present, and mitotic figures were increased in number (Fig. 5). Also seen were occasional microcysts (Fig. 6), and areas of hyalinized fibrosis (Fig. 7). Immunohistochemistry positively marked the spindled cells for vimentin and actin. Stains were negative for desmin, S100 and cytokeratin.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Diagnosis: “Nodular Fasciitis with Giant Cells, Posterior Left Thighâ€
Marnelli A. Bautista, MD, and Donald R. Chase, MD
Department of Pathology and Human Anatomy, Loma Linda University and Medical Center, Loma Linda, CA
California Tumor Tissue Registry, Loma Linda, CA
Discussion: Nodular fasciitis (NF) is a benign, reactive process consisting of myofibroblasts and fibroblasts. In 1955, Kornwaler and colleagues first described the lesion as “subcutaneous pseudosarcomatous fibromatosis†due to its rapid growth, areas with higher cellularity, and presence of mitotic figures, making it the most common benign mesenchymal lesion misdiagnosed as sarcoma. Commonly involved sites are the upper extremities, trunk, chest and back; however, it can also occur in the extremities, perhaps due to trauma. NF is hypothesized to be initiated by injury largely because it resembles organizing granulation tissue typically encountered in reactive or reparative processes. This lesion arises most commonly in adults 20-40 years of age, although the literature reports that about 10% of the cases are diagnosed in children and an additional 10% arise in patients more than 60 years.
Key features of NF include:
• Small size, generally less than 2 cm, with a well-circumscribed periphery
• Rapid initial growth, but regression over time
• Regional heterogeneity with prominent myxoid background and areas with hyalinized fibrosis
• Haphazardly arranged, uniform myofibroblasts and fibroblasts, with tissue culture growth-pattern or “torn tissue paper†appearance
• Increased mitotic figures
Correlation between the microscopic features and the duration of the lesion has been established. The acute phase usually has a mucoid appearance with less cellularity. Over a short period of time, an increasing myxoid matrix causes the lesion to expand. Then, a myofibroblastic proliferation occurs, which microscopically results in an appearance of increased cellularity. Long-standing processes sometimes referred to as the “resolving stageâ€, tend to have hyalinized fibrosis. This “sclerosis†seemingly reflects involution of the nodule. Microcysts may also be seen in this final stage. The eventual appearance may be that of a “dermatofibroma†or “fibroma.â€
The myofibroblasts in NF are randomly arranged, being separated by the expanding mass of mucopolysaccharide matrix and the proliferation of capillaries. At this time the process may assume a feathery or fibrillary appearance, sometimes likened to as a “tissue culture-like appearance.†Reflecting the rapid expansion of the nodule, mitotic figures are numerous but atypical forms are rarely seen. Occasional red cells and lymphocytes can be identified. A few giant cells may also be present, but are very uncommon. Okoye and colleagues demonstrated that the giant cells in NF are likely related to myofibroblasts by virtue of their similarity in ultrastructural and immunohistochemical characteristics. This finding further supports the hypothesized reparative process in NF, similar to the response generated by injury.
Three described “classic†subtypes are:
• Subcutaneous NF – a well-defined nodule; most common among the three subtypes
• Intramuscular NF – slightly larger than the other two subtypes
• Fascial NF – less delineated due to its propensity to grow along septa producing a stellate pattern
Other benign variants of NF include:
• Intravascular fasciitis is a rare type (<3% of NF cases), most commonly found in younger patients. It is characterized by slow growth and mainly involves small and medium sized vessels, which could be mistaken for a thrombus. It has a less myxomatous background and tends to have more giant cells. • Cranial fasciitis primarily presents during the first year of life and is believed to be associated with birth trauma. This well-demarcated lesion is characterized by rapid growth, which may involve the dura & meninges. It consists of prominent myxoid and hyalinized matrix, and may occasionally have focal bone metaplasia. • Ossifying fasciitis is a less well-circumscribed nodule, with numerous foci of bone metaplasia. Differential diagnoses include: • Proliferative fasciitis (PF) - may be distinguished by characteristic large cells appearing similar to ganglion cells and having one or two large, basophilic, peripherally located nuclei. The clinical course, however, is the same as NF. • Giant Cell Tumor of Tendon Sheath (GCTTS) - mainly occurs adjacent to the interphalangeal joints of the fingers, a very unusual location for NF. Histologically, GCTTS is not characterized by a myxoid matrix, and has a lobular appearance. Giant cells are usually prominent, and have many more nuclei than are seen NF. • Fibromatosis - typically is a poorly demarcated lesion which infiltrates the surrounding subcutaneous tissue. It consists of long slender-shaped fibroblasts and lacks the characteristic mucoid background. Giant cells are never seen. • Myxoma - may mimic the acute phase of nodular fasciitis with expansion of myxoid matrix, but no proliferation of myofibroblasts is seen. The process does not regress. Unlike NF, the process is virtually non-vascular. • Fibrous histiocytoma - may resemble the cellular or early resolving phase of nodular fasciitis. This entity consists of spindle cells and polygonal histiocytes usually arranged in a cartwheel or storiform pattern. NF, however, may ultimately mimic this entity in its end “resolution†state, • Fibrosarcoma (FS) - can be differentiated from NF by the organized fibroblasts arranged in interweaving bundles resulting in a herringbone pattern. Although it shares with NF a high mitotic rate, FS does not show the tissue-paper myxoid phenomenon. In general, complete surgical excision is curative, and this lesion rarely recurs once removed. The literature reports about 1% recurrence rate. Reported exceptions include an article by Thompson who reported approximately 9% recurrence of external ear lesions. Another article documented 13% recurrence rate, although upon re-review, all recurrent lesions were later re-classified as entities other than NF. In general, NF is felt to be a non-recurrent reparative process. Suggested Reading:
Bernstein KE; Lattes R. Nodular (Pseudosarcomatous) Fasciitis, A Nonrecurrent Lesion: Clinicopathologic Study of 134 Cases. Cancer 49(8): 1668-78, 1982.
Okoye MI; Watanabe I. Ultrastructural and Immunohistochemical Investigations of the Giant Cells in Nodular Fasciitis. J Natl Med Assoc 80 (7): 770-5, 1988.
Akihiro Y; Hideki O. Nodular Fasciitis with Degeneration and Regression. J Craniofac Surg 19(4): 1167-70, 2008.
Lenyoun EH; Wu JK; Ebert B; Lieberman B. Rapidly Growing Nodular Fasciitis in the Cheek of an Infant: Case Report of a Rare Presentation. Eplasty 8: e30, 2008.
Thompson LD; Fanburg-Smith JC; Wenig BM. Nodular Fasciitis of the External Ear Region: A Clinicopathologic Study of 50 Cases. Ann Diagn Pathol 5(4): 191-8, 2001.
Weiss S, Goldblum J. Enzinger & Weiss’s Soft Tissue Tumors (5th ed). Philadelphia: Mosby/Elsevier Inc. 177-92, 2008.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}