History: A sixty-one year old woman underwent a hand-assisted laparoscopic nephrectomy. The 256.0 gram, 12.0 x 6.5 x 2.5 cm left kidney contained a single 4.3 x 4.0 x 3.2 cm homogeneous tan-white, pseudo-encapsulated mass in the mid ante-hilar pole. The mass was adjacent to but did not penetrate the renal capsule. No tumor involvement of the perirenal adipose, hilar vessels, renal pelvis or ureter was noted. The residual renal parenchyma had a normal red-brown appearance and showed a distinct cortico-medullary junction. No other lesions were identified.
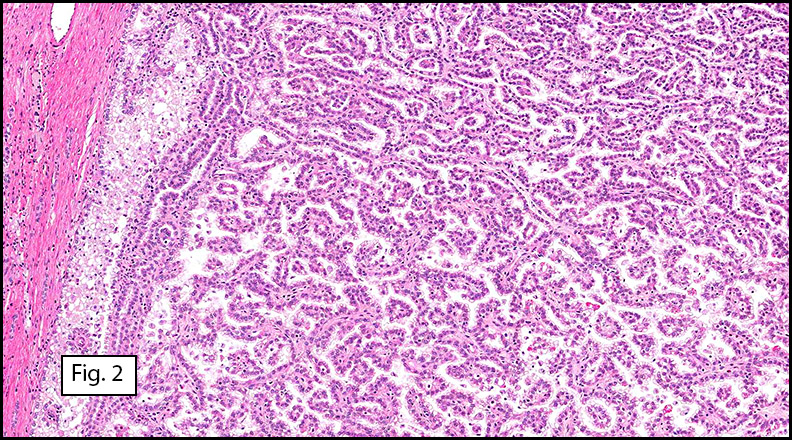
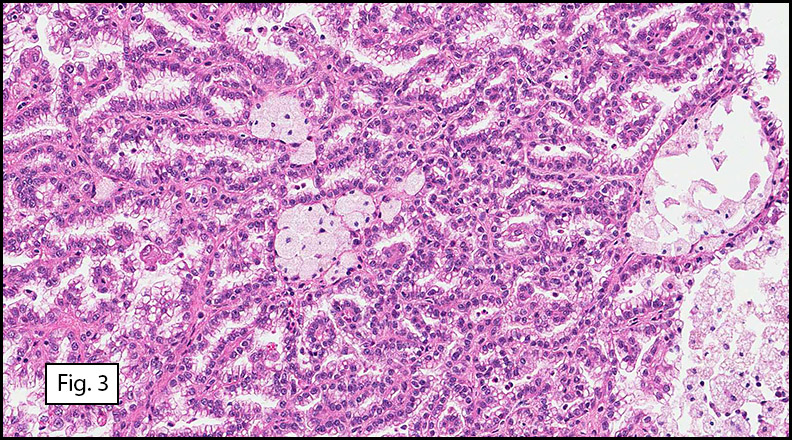
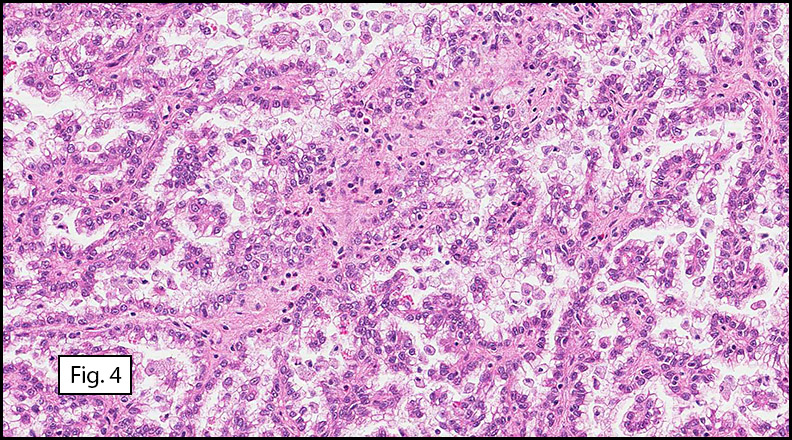
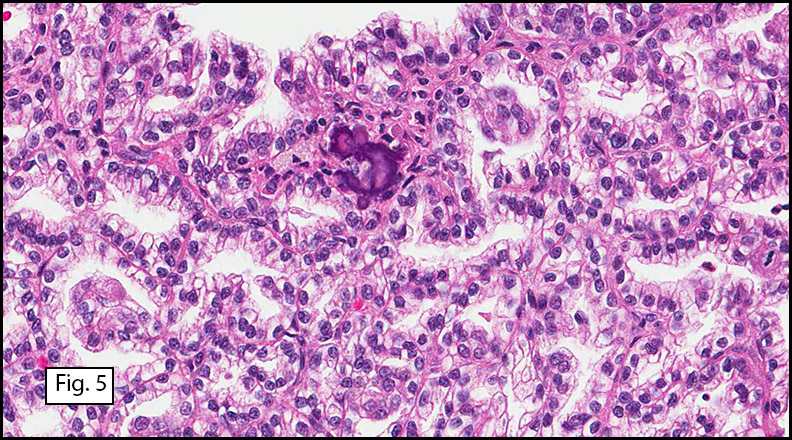
Microscopically, the well-circumscribed tumor approached but did not extend through the renal capsule (Fig. 1). A distinctive tubulo-papillary architecture was observed (Fig. 2), with fibrovascular cores lined by a single layer of tumor cells. The tumor cells had moderate to focally abundant basophilic cytoplasm. The low-grade nuclei displayed open chromatin and rare nucleoli, consistent with Fuhrman Nuclear Grade 2. Occasional foamy histiocytes (Fig. 3), clear cells (Fig. 4), and psammoma bodies were seen (Fig. 5). Sarcomatoid features, tumor necrosis, and lymphovascular invasion were absent. The adjacent non-neoplastic kidney demonstrated chronic inflammation and hyalinized glomeruli, consistent with mild benign nephrosclerosis.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Diagnosis: “Papillary Renal Cell Carcinoma, Type 1 (Basophilic)â€
Rachel Conrad, MD1; Pamela Boswell, DO2; Manju Aron, MD3; Mahul Amin, MD3; Donald R. Chase, MD1,4.
1. Department of Pathology and Human Anatomy, Loma Linda University and
Loma Linda Medical Center, Loma Linda, California
2. Scripps Clinic Medical Laboratories, La Jolla, California
3. Cedars Sinai Medical Center, Los Angeles, California
4. California Tumor Tissue Registry, Loma Linda, California
Discussion: Papillary renal cell carcinoma is a relatively rare type of renal cell carcinoma with unique morphologic, immunohistochemical, and genetic features. It tends to occur in the sixth to seventh decades with a male predominance (3:1).
Papillary renal cell carcinoma has been associated with several different clinical syndromes. Hereditary/familial papillary renal cell carcinoma (7q31 c-MET mutation) results in multiple bilateral type 1 papillary renal tumors. Hereditary leiomyomatosis and renal cell cancer (1q42-43 mutation) leads to a solitary unilateral type 2 papillary renal cell carcinoma, usually around age thirty, along with cutaneous and uterine leiomyomas and leiomyosarcomas. Birt-Hogg-Dube syndrome (17p11.2 mutation) has multiple chromophobe and oncocytoma renal cell tumors with rare clear cell or papillary renal cell carcinomas. Fibrofolliculomas, lung cysts, and a history of spontaneous pneumothorax are frequently noted. Familial paraganglioma syndrome displays multiple renal tumors with benign and malignant paragangliomas and papillary thyroid carcinoma. PTEN hamartoma tumor syndrome results in unifocal papillary RCC and chromophobe RCC with gastrointestinal polyposis, tumors of the breast, thyroid and uterus, and frequently, lipomas and trichilemmomas.
The gross appearance of papillary renal cell carcinoma consists of a well-circumscribed nodule with a fibrous pseudocapsule. Lesions are often bilateral and multifocal, and hypovascularity may be seen on radiographic studies. Cross-sectioning reveals a yellow-brown surface with regions of cystic degeneration, hemorrhage, and necrosis.
Microscopically, the tumors contain fibrovascular cores lined by neoplastic cells containing foamy histiocytes and occasional neutrophils. Solid-papillary and tubulopapillary growth patterns may also be observed. Sarcomatoid dedifferentiation is seen in five percent of papillary renal cell carcinomas and confers a poor prognosis. Additional findings may include stromal and intracellular hemosiderin deposition, occasional psammoma bodies, necrosis, and clear cell change in areas of hemorrhagic degeneration.
Papillary renal cell carcinoma has been classified by Delahunt and Eble (1997) into two subtypes. Type 1 (also known as “basophilicâ€) shows a single layer of cells with scant basophilic-to-amphophilic finely granular cytoplasm and low-grade nuclei arranged along the papillary basement membrane. Type 2 shows pseudostratified cells with abundant eosinophilic granular cytoplasm and large nuclei displaying a higher Fuhrman grade. As expected, the higher grade tumors have a worse prognosis.
Immunohistochemical staining shows membranous positivity for CK7, with stronger expression in Type 1 papillary renal cell carcinoma. A granular cytoplasmic pattern is seen for alpha methylacyl CoA-racemase (AMACR). Other positive stains include AE1/AE3, CAM5.2, EMA, CD10, PAX2, PAX8, RCC, and PN15/gp200. Negative staining is seen for CAIX, Cathepsin-K, TFE3, and 34-beta-E12.
Cytogenetic studies often show trisomy 7, trisomy 17, and loss of Y without abnormalities of 3p. Additional genetic anomalies may be observed.
The recommended therapy for papillary renal cell carcinoma is complete extirpation. The overall prognosis is better than for conventional renal cell carcinoma, with a 5 year survival rate of 45-70%. Survival drops to 15-20% if extension to the renal vein or perinephric fat is observed. Metastases are frequently seen in tumors larger than 3 cm and tend to involve the lungs, lymph nodes, bones, and liver.
The differential diagnosis of papillary renal cell carcinoma includes metanephric adenoma, conventional clear cell renal cell carcinoma with papillary foci, clear cell papillary renal cell carcinoma, oncocytic papillary renal cell carcinoma, Xp11 translocation renal cell carcinoma, collecting duct carcinoma, and mucinous tubular and spindle cell carcinoma of the kidney.
1. Metanephric adenoma tends to occur in young to middle aged females. The microscopic appearance is consistent with developing metanephric tubular epithelium and displays tiny tubules and papillae with bland nuclei and scant stroma. This lesion is usually small in size and benign in nature. Immunostaining shows rare focal CK 7 positivity and negativity for EMA and AE1/AE3.
2. Conventional clear cell renal cell carcinoma with papillary foci is fairly common. It may be distinguished from papillary renal cell carcinoma by its hypervascularity on radiographic imaging, golden yellow color on sectioning, and solid or acinar architecture with clear-to-eosinophilic cells separated by thin vascular septae. True papillae are rarely seen; instead, small papillations protrude into necrotic or cystic spaces. Chromosome studies frequently show deletion of the von Hippel-Lindau gene (3p), and inactivation of this gene can be detected by diffuse strong membranous staining for CAIX. Negative immunohistochemical staining is observed with CK7 and AMACR.
3. Clear cell papillary renal cell carcinoma is a small indolent tumor associated with end-stage renal disease. Microscopically, the papillary fronds are lined by clear cells with low Fuhrman nuclear grade and characteristic subnuclear vacuoles. Smooth muscle metaplasia and cystic changes are common. No hemosiderin deposition, foamy histiocytes, or psammoma bodies are seen. Immunohistochemistry shows a cup-like pattern for CAIX and CK7, with patchy positivity for 34-beta-E12; CD10 and AMACR are negative.
4. Oncocytic renal cell carcinoma with papillary features has a distinctive mahogany-brown color with a central scar on sectioning. Microscopic features include abundant granular eosinophilic cytoplasm and a linear arrangement of low-grade apical nuclei. Pseudostratification is absent, and papillary architecture is focal, not diffuse. IHC staining is positive for parvalbumin and negative for RCC and CK7.
5. Xp11 translocation renal cell carcinoma is the most common pediatric renal cell carcinoma subtype and may be associated with previous chemotherapy. Extensive psammomatous calcification may be seen radiographically and microscopically. Papillary architecture with clear-to-eosinophilic cells and vesicular high-grade nuclei is common, but cystic, solid, or nested architecture can also be seen. Immunohistochemical staining is positive for Cathepsin-K, TFE3, and PAX8, with focal staining for melanocytic markers. Weak expression of cytokeratins and epithelial markers may be seen. Cytogenetic studies often show translocation of ASPL and TFE3, t(X;17)(p11.2q25).
6. Collecting duct carcinoma tends to display prominent desmoplasia, frequent invasion, and high grade nuclear features. IHC stains are negative for CD10, AMACR, and PN15/gp200.
7. Mucinous tubular and spindle cell carcinoma of the kidney can be distinguished due to its mucinous stroma and overall architectural pattern. It may share focal papillary growth, mucin production, foam cells, and AMACR positivity with papillary renal cell carcinoma, but it will lack the classic diffuse papillary pattern.
Due to its better prognosis, associated familial syndromes, and tendency toward multiple bilateral hypovascular tumors, papillary renal cell carcinoma is important to separate from other lesions with papillary architecture. Characteristic microscopic traits include true fibrovascular cores containing foamy histiocytes and hemosiderin. Immunohistochemical stains (CK7, AMACR) and cytogenetic studies (trisomy 7/17, loss of Y) can be useful in challenging cases. Treatment consists of surgical excision and careful assessment for metastases. The astute observer will be alert to the unique aspects of this rare tumor.
Suggested Reading:
1. Delahunt B, Eble JN. Papillary renal cell carcinoma. In: Eble JN, Sauter G, Epstein JI, Sesterhenn IA, eds. World Health Organization Classification of Tumours. Pathology and Genetics of Tumors of the Urinary System and Male Genital Organs. Lyon, France: IARC Press, 2004:15-22.
2. Merino MJ, Eccles DM, Linehan WM, et al. Familial renal cell carcinoma. In: Eble JN, Sauter G, Epstein JI, Sesterhenn IA, eds. World Health Organization Classification of Tumours. Pathology and Genetics of Tumors of the Urinary System and Male Genital Organs. Lyon, France: IARC Press, 2004:15-22.
3. Mester JL, Zhou M, Prescott N, Eng C. Papillary renal cell carcinoma is associated with PTEN hamartoma tumor syndrome. J Urol. 2012; 79(5):1187.e1-7
4. Sukov WR, Lohse CM, Leibovich BC, Thompson RH, Cheville JC. Clinical and pathological features associated with prognosis in patients with papillary renal cell carcinoma. J Urol, 2012; 187(1):54-59.
5. Ross H, Martignoni G, Argani P. Renal cell carcinoma with clear cell and papillary features. Arch Pathol Lab Med. 2012;136(4):391-9.
6. Rosai J. Rosai and Ackerman’s Surgical Pathology. China: Mosby, 2004:1251-1264.
7. Delahunt B, Eble JN. Papillary renal cell carcinoma: a clinicopathologic and immunohistochemical study of 105 tumors. Mod Pathol. 1997;10:537.
8. Murphy WM, Grignon DJ, Perlman EJ. AFIP Atlas of Tumor Pathology Series 4: Tumors of the Kidney, Bladder, and Related Urinary Structures. Washington, D.C.: American Registry of Pathology, 2004.