History: A 36 year old woman presented with early satiety, recent weight gain and dyspnea on exertion. She was found to have a palpable left upper quadrant mass, which upon MRI was mostly solid. The excised pancreatic tumor was 23 by 17 cm, well circumscribed and surrounded by a thin capsule or pseudocapsule. Sectioning showed it be mottled gray and brown, friable, and to have prominent fibrous septa.
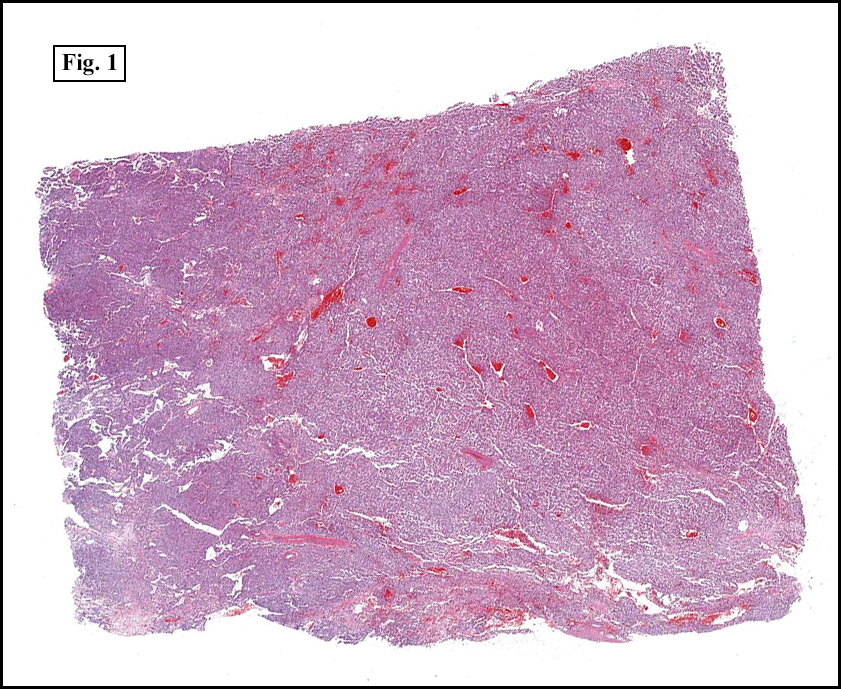
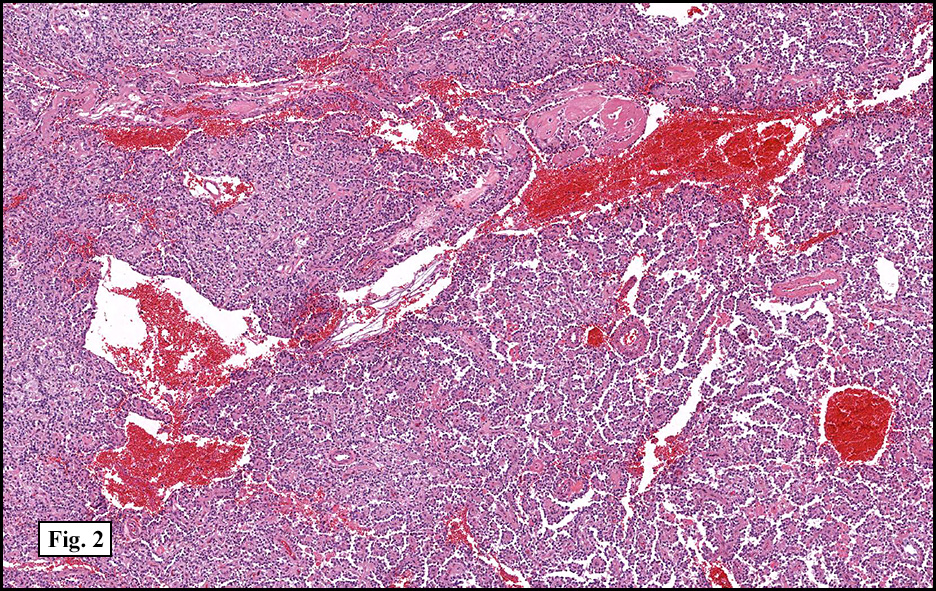
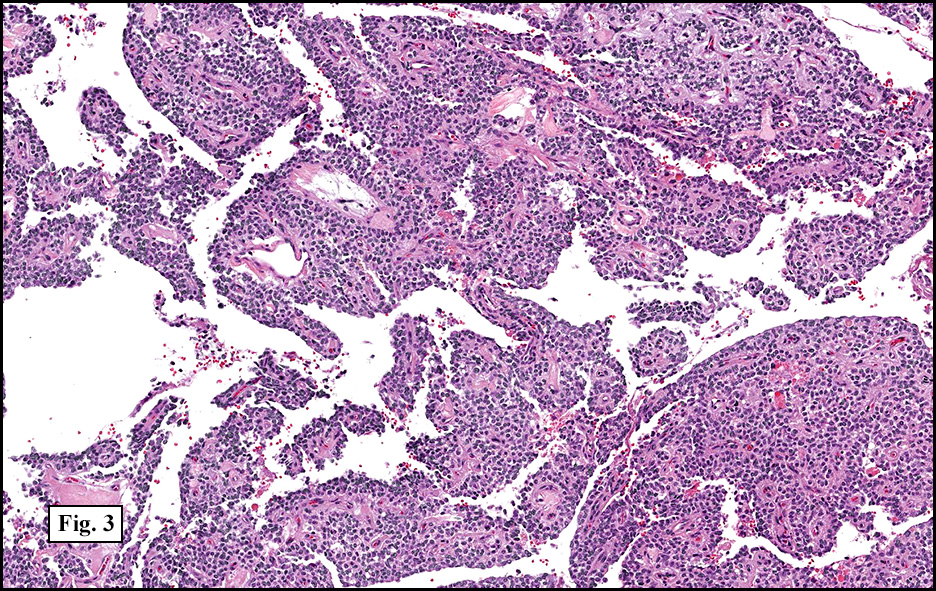
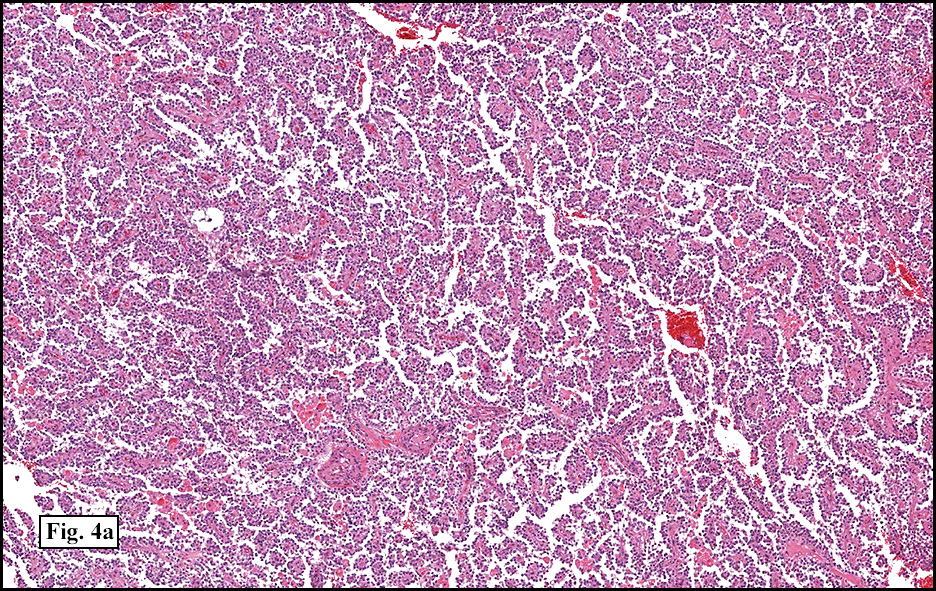
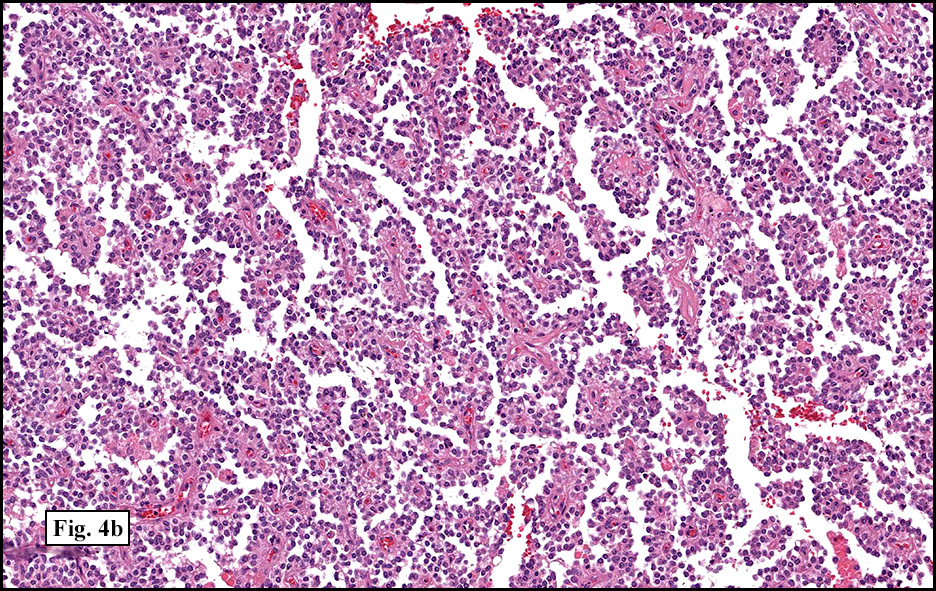
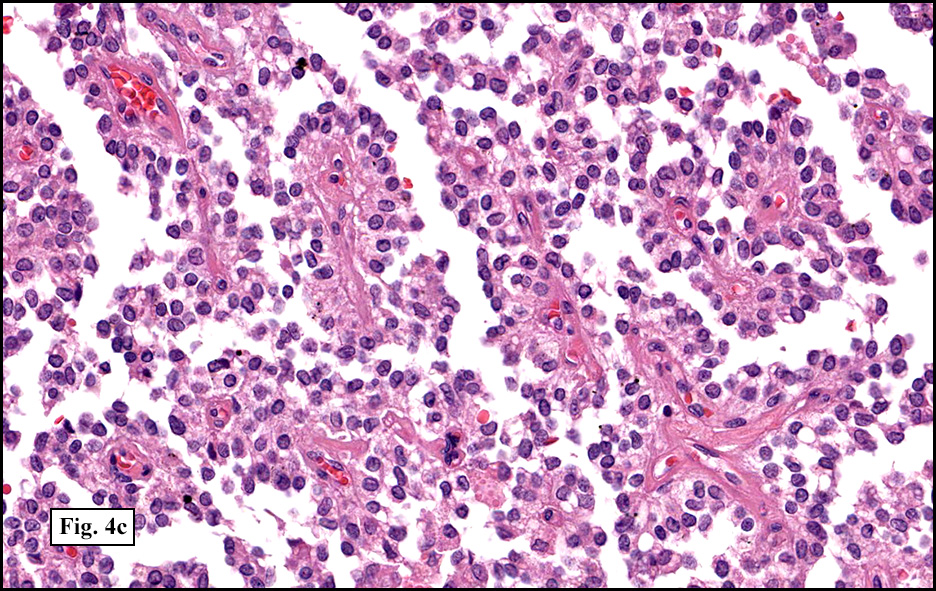
Microscopically, tumor cells were small, rounded and uniform. They lacked significant pleomorphism, atypism, or mitotic figures. The tumor was mostly solid, without significant cystic change (Fig. 1). Vascularity was prominent and included medium-sized vessels many of which contained thrombi (Fig. 2). Necrosis, however, was scant. Although a sinusoidal growth pattern was focally present (Fig. 3), most of the tumor pattern was “papillary†(Figs. 4a, 4b, 4c).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Diagnosis: “Solid Pseudopapillary Tumor of the Pancreasâ€
Cody S. Carter, MSIV, Donald R. Chase, M.D.
Department of Pathology and Human Anatomy, Loma Linda University and Medical Center, Loma Linda, California
California Tumor Tissue Registry, Loma Linda, California
Discussion: Solid pseudopapillary tumor of the pancreas (SPT) is a rare tumor, previously known as â€solid cysticâ€, “papillary cysticâ€, and “solid and papillary epithelial neoplasmâ€. About 95% of cases occur in women, and the average age at presentation is in the mid-20s. The tumor does not spare any age group, although it rarely occurs prior to adolescence. The most common presenting symptoms are abdominal pain followed by a palpable abdominal mass. Other symptoms include nausea, vomiting, fever, weight loss, and jaundice. Up to 15% cases are asymptomatic, detected incidentally on imaging studies. Because of this, the tumors can reach very large sizes with the average size being 7 cm at the time of resection.
Grossly, SPT is usually a well circumscribed mass with a yellow-brown, hemorrhagic cut surface. The tumors are solid but undergo cystic degeneration with increased size, and as a result are often soft and friable. Because of the tendency for the tumors to go undetected until reaching a large size, there may be a significant cystic component, sometimes mimicking a pancreatic pseudocyst. These cysts often contain hemorrhagic fluid.
Microscopically, SPT demonstrates solid nests of largely uniform epithelioid cells intermixed with small blood vessels. The characteristic pseudopapillae are formed as the cells further away from the blood vessels degenerate and become dyscohesive. Microcystic and cyst-like spaces are often present and are formed by degenerative changes. The tumor cells exhibit a moderate amount of delicate, eosinophilic cytoplasm. The cytoplasm often contains diastase-resistant PAS-positive eosinophilic hyaline globules. These globules are composed of alpha-1 antitrypsin. Nuclei are round to ovoid and frequently feature grooves and membrane irregularities, but do not usually exhibit pleomorphism and mitotic figures.
Cytology is useful to gain a preoperative diagnosis and direct surgical intervention, and is often necessary because of a lack of specific radiographic features that differentiate this entity from other cystic lesions. FNA biopsies usually demonstrate highly cellular material with branching, delicate papillary structures consisting of fibrovascular cores covered by one to a few cell layers. Tumor cells are of uniform size and shape, with pale ill-defined cytoplasm. The round or ovoid nuclei feature finely granular chromatin which may have grooves. Hyaline globules are easily identified by their red color on Giemsa stain. Foamy histiocytes are also present in varying quantities.
Of particular interest is the lack of a consensus on the cellular origin of SPT despite extensive immunohistochemical and genetic studies. Immunohistochemistry, while exceedingly helpful in other diagnoses, have not been particularly useful in SPT. The tumor cells are diffusely positive for vimentin and galectin-3, and most tumors demonstrate positivity for neuron specific enolase, α-1-antitrypsin, and CD56, while few cases show distinct immunoreactivity with cytokeratin, synaptophysin, chromogranin, trypsin, and S-100 protein. The weak reactivity for these markers provides no convincing evidence for ductal, acinar, or endocrine differentiation. Also of interest is the occurrence of this tumor almost exclusively in women. This has led many experts to believe that hormonal signaling plays a major role in tumorigenesis, and indeed, a high proportion of tumors have been shown to be immunoreactive to both the progesterone receptor and estrogen receptor-β. However, the similarity between affected men and women in regards to growth pattern and affected age groups is puzzling if hormone receptors prove to be involved in the evolution of this neoplasm. Despite this, hormonal manipulation may be indicated.
Molecular analysis of SPT demonstrates mutations in the β-catenin gene (CTNNB1) on chromosome 3p in more than 90% of tested cases. These mutations result in the inactivation of one of the glycogen synthase kinase-3β phosphorylation sites on the β-catenin protein, which inhibits the degradation of β-catenin. This lack of degradation leads to a buildup of β-catenin in the cytoplasm and nucleus of the tumor cells, resulting in a change in staining pattern for β-catenin protein antibodies. Whereas normal cells have a membranous staining pattern for β-catenin, tumor cells of SPT have an abnormal nuclear staining pattern. The consistency of this nuclear staining makes this marker a diagnostically important adjunct to other immunostains when histology cannot be confidently relied upon. The activation of the β-catenin protein also stimulates transcription of a number of genes that promote neoplasia including cyclin D1 and c-myc. Another effect of the mutation is an interaction between the β-catenin protein with E-cadherin, resulting in a loss of the membranous labeling for the extracellular domain of E-cadherin that is normally seen in non-neoplastic pancreas. Due to the integral role that E-cadherin plays in cell-cell junctions, this disruption has been postulated as the reason for the dyscohesion seen between tumor cells and thus the cystic degeneration often seen grossly.
SPT is a malignant tumor with low metastatic potential. It has a 5-year survival rate of 95%, and patients are almost always cured by surgical resection. Metastases rarely occur after resection if not already present at the time of diagnosis, and even with metastases, patients may often live for many years. As a result, local metastases and recurrence are not absolute contraindications to resection. Some patients with metastatic disease have survived more than 10 years after surgery, and in contrast to other pancreatic neoplasms, tumor debulking is recommended. In rare cases of unresectable SPTs, radiotherapy has been shown to be effective.
Differential diagnosis:
• Acinar cell carcinoma typically affects elderly patients and only rarely have cystic variants been seen. Similar to SPT, these tumors consist of uniform cells with granular cytoplasm that can stain positively with diastase-resistant PAS. However, the cells are arranged in acinar and trabecular patterns, and immunohistochemistry demonstrates widespread positivity for cytokeratin, trypsin and chymotrypsin, while vimentin is usually negative.
• Pancreatoblastoma typically occurs in the first decade and is characterized by epithelial cells organized in sheets, acini and squamoid corpuscles, surrounded by a fibrous stroma. In contrast to SPT, it is negative for vimentin and strongly positive for cytokeratin and trypsin.
• Pancreatic endocrine neoplasm (PEN) shares similar cellular characteristics with SPT, featuring bland, uniform cells with finely granular cytoplasm and ovoid nuclei. These cells are typically in solid and trabecular patterns without papillary structures. The immunophenotype of PEN is consistently reactive for cytokeratin and neuroendocrine markers such as chromogranin and synaptophysin. Vimentin, if present, is usually weak and focal.
Suggested Reading:
Adams AL, Siegal GP, Jhala NC. Solid pseudopapillary tumor of the pancreas. A review of salient clinical and pathologic Features. Adv Anat Pathol. 2008 Jan; 15(1):39-45.
Chanjuan S, Daniels JA, Hruban RH. Molecular characterization of pancreatic neoplasms. Adv Anat Pathol. 2008 Jul; 15(4):185-95.
Papavramidis T, Papavramidis S. Solid pseudopapillary tumors of the pancreas: Review of 718 patients reported in English literature. J Am Coll Surg. 2005 Jun; 200(6):965-972.
Adsay VN. Cystic neoplasia of the pancreas: pathology and biology. J Gastrointest Surg. 2008 Mar; 12(3):401-404.
Klimstra DS, Wenig BM, Heffess CS. Solid-pseudopapillary tumor of the pancreas: a typically cystic carcinoma of low malignant potential. Semin Diagn Pathol. 2000 Feb; 17(1):66-80.
Yu PF, Hu ZH, Wang XB, Guo JM, Cheng XD, Zhang, YL, Xu Q. Solid pseudopapillary tumor of the pancreas: A review of 553 cases in Chinese literature. World J Gastroenterol 2010 Mar;16(10)1209-1214.
Pettinato G, Di Vizio D, Manivel JC, Pambuccian SE, Somma P, Insabato L. Solid-pseudopapillary tumor of the pancreas: A neoplasm with distinct and highly characteristic cytological features. Diagnostic Cytopathology 2002 Dec; 27(6): 325-334.
Geers C, Pierre M, Jean-Francois G, Birgit W, Pierre D, Jacques R, Christine S. Solid and pseudopapillary tumor of the pancreas—review and new insights into pathogenesis. Am J Surg Pathol. 2006 Oct; 30(10): 1243-1249.
Odze R, Goldblum J. Surgical Pathology of the GI Tract, Liver, Biliary Tract, and Pancreas (2nd ed). Philadelphia: Saunders/Elsevier Inc. 944-48, 2009.