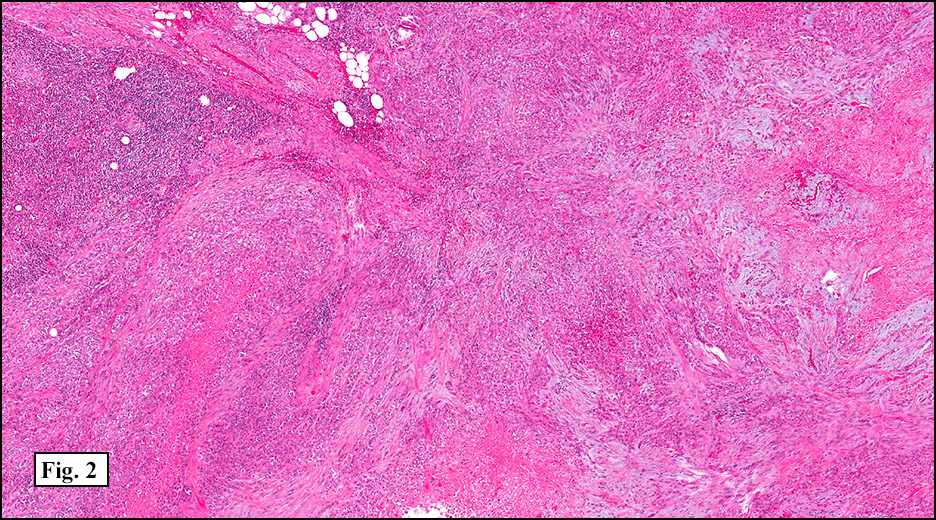
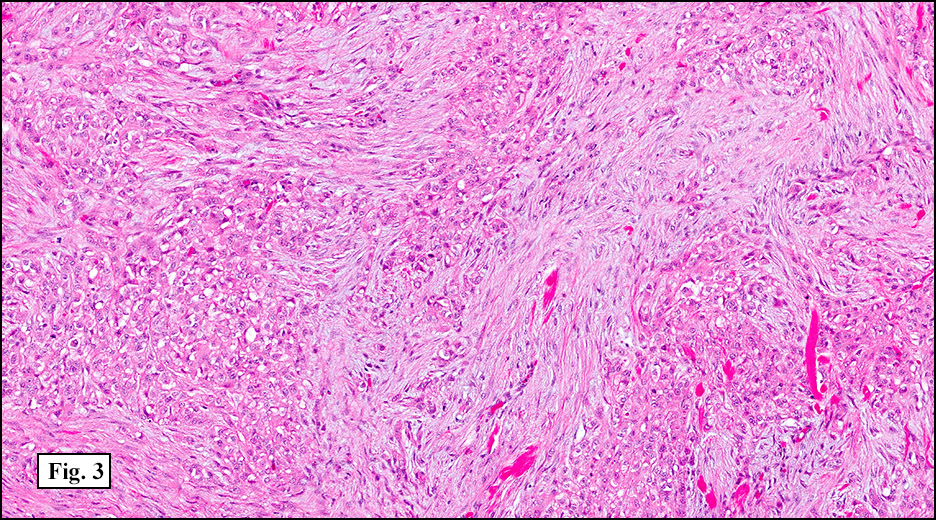
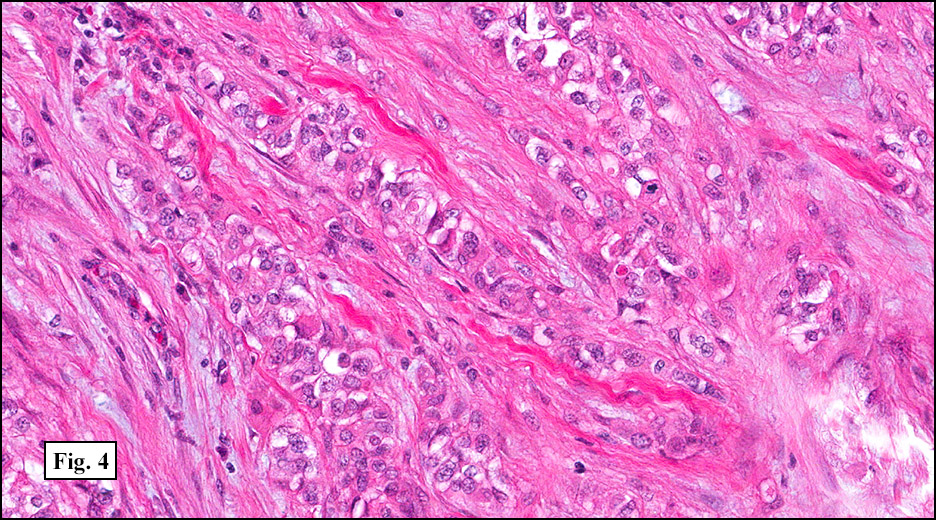
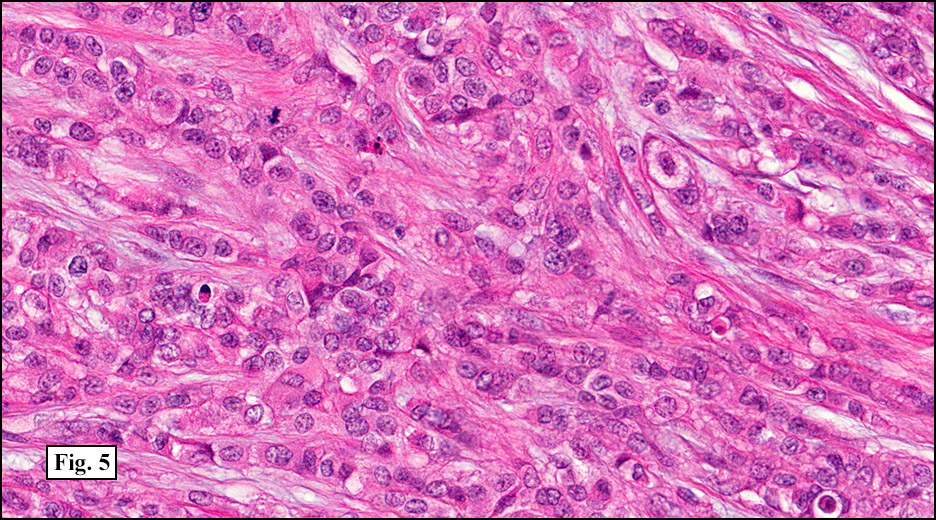
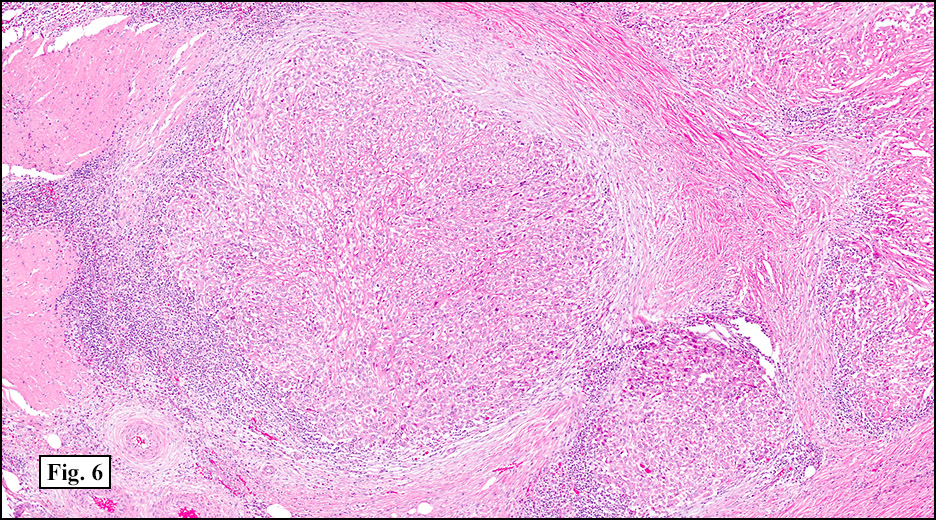
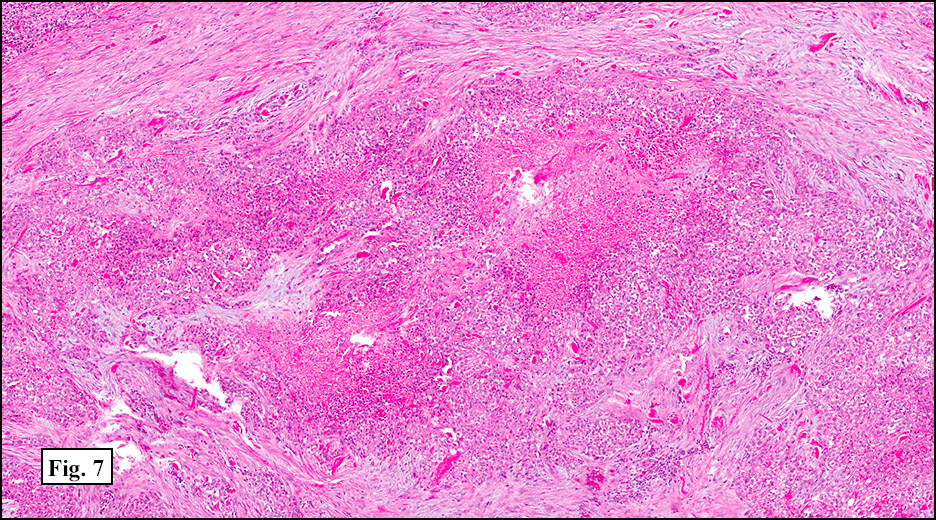
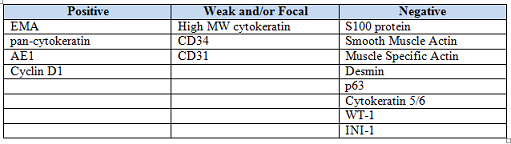
History: A 33-year-old man underwent resection of a 3.5 x 2.0 cm, inguinal, gray-white tumor with regional necrosis (Fig. 1). The neoplastic cells were eosinophilic and mostly epithelioid or spindloid (Figs. 2, 3). Their nuclei were vesicular and generally had one to two small nucleoli and showed occasional mitotic figures (Figs. 4, 5). They focally formed granuloma-like patterns with merging central cores of necrosis mimicking “caseous necrosis†seen in various infectious processes (Figs. 6, 7). Minimal pleomorphism was present. Neither glandular structures nor squamous differentiation was encountered. Immunostains were interpreted as follows:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Diagnosis: Epithelioid Sarcoma, Large Cell (“Proximalâ€, “Rhabdoidâ€) Variant
Rachel Conrad, MD1; Scott G. Silveira, MD2; Donald R. Chase, MD1,3
1. Department of Pathology and Human Anatomy, Loma Linda University and Loma
Linda Medical Center, Loma Linda, California
2. Department of Pathology, Oakwood Hospital and Medical Center, Dearborn,
Michigan
3. California Tumor Tissue Registry, Loma Linda, California
Discussion: First described by Franz Enzinger in 1970, epithelioid sarcoma (ES) has been accepted as a unique soft tissue sarcoma which typically pursues an indolent but relentless course with numerous recurrences. It generally occurs in young adults (10-35 years of age), but may occur in the very young as well as in the elderly. Males are affected twice as often as females.
The distal upper extremities are the most common sites, followed by the distal lower extremities, the upper proximal extremities, the trunk, and the head and neck. Case reports have also described rare lesions in the spine, gingiva, orbit, tongue, esophagus, jejunum, iliac bone, and intra-articular joints. Unlike most sarcomas, ES has a tendency to initially metastasize to regional lymph nodes. Lung metastases are common, and the tumor has a predilection for scalp and brain metastases.
Most ES’s present as painless, slow-growing, palpable firm masses in the soft tissue or dermis. Although the average size is 3-6 cm, deeper lesions are likely to be larger, multinodular, and attached to tendons or nerves which may result in localized neural symptoms including carpal tunnel syndrome or median nerve palsy. Antecedent trauma has been reported in up to 20% of cases. Rare reports also describe ES occurring in conjunction with squamous cell carcinoma, lung adenocarcinoma, neurofibromatosis type 2, acute lymphoblastic leukemia, and exposure to hydrazine fuel or Agent Orange. Elevated serum CA-125 has been reported in a few cases and may prove useful in tracking disease progression.
Radiographic findings often reveal a nonspecific solid or cystic multilobulated lesion with small speckled calcifications. Adjacent bone erosion and thinning may be seen, but invasive osseous destruction is rare.
The usual gross appearance of ES is of firm, non-encapsulated tan-white nodules with indistinct, infiltrating margins. Superficial tumors may have skin ulceration or sinus tract formation. Hemorrhage and/or necrosis is usually present.
As originally described, the tumor is nodular with central degenerative necrosis. The tumor is comprised of eosinophilic epithelioid cells admixed with polygonal to plump spindloid cells and dense intercellular collagen. Minimal pleomorphism is seen. Mitotic figures range from 1-14/10 HPF. Vascular invasion is much more common than neural invasion. Some tumors are lymphotrophic, showing a chronic inflammatory cell infiltrate around their peripheries. Calcification and osseous metaplasia may be present, but cartilaginous change is rare.
The classically defined subtypes, “necrobiotic†and “solid,†were extensively studied in the 1985 paper by Chase and Enzinger. Additional variants such as “fibroticâ€, “angiomatoid†and “large cell†were also noted at that time, and have received substantial recent attention
1. The necrobiotic (“granulomatousâ€) growth pattern consists of coalescent cystic nodules lined by undulating ribbons of polygonal and spindled cells. The spaces are filled with necrotic debris or hyalinized collagen. The epithelial-appearing cells are mostly centrally located and the spindled elements are toward the periphery. No abrupt transition is seen. Nuclei are rounded and usually show small nucleoli. Minimal pleomorphism is seen.
2. The solid (“nodularâ€, “lobularâ€) growth pattern has a multinodular lobular growth pattern with polygonal and spindled cells similar to those seen in the necrobiotic form; however, no significant necrosis is seen. Nuclei typically are eccentrically located, pushed aside by the intermediate filaments vimentin and cytokeratin. As with the necrobiotic variant, the cells have distinct cytoplasmic borders.
3. Unlike the “classic†necrobiotic and solid growth patterns, the fibrotic (“fibroma-likeâ€) variant of ES consists mostly of spindled cells, with a lesser epithelioid cell component. Cells reside in a fibrous stroma and show a vague storiform growth pattern resembling a cellular fibrous histiocytoma.
4. The large cell (“proximal,†“rhabdoidâ€) variant usually occurs in deep tissues of limb girdles or buttocks, vulva and perineum, and consists of polygonal cells which are larger and more pleomorphic than the other variants of ES. The cells have increased numbers of mitoses. Some cells may resemble rhabdomyoblasts. This variant may express smooth muscle actin; however, desmin positivity is lacking. The rhabdoid phenotype is more commonly found in tumors that are “proximal†or “truncal†but is not unique to the proximal form of epithelioid sarcoma. Since similar cytomorphology may be seen in other tumors (synovial sarcoma, extraskeletal myxoid chondrosarcoma, malignant mesothelioma, bladder carcinomas), it may be viewed simply as a pattern, similar to the patterns of pleomorphic “MFH†and HPC. In view of the increased risk of recurrence and metastasis that are associated with “rhabdoid cells†in general, and with an ES in a proximal location, this variant may have a faster progression.
5. The angiomatoid type is the rarest of the ES variants. Histologically the tumors show cystic, blood-filled spaces bordered by epithelioid and spindled cells. An endothelial lining to the spaces is not seen; neither is vasoformative activity observed. CD34 may be positive in up to 50% of the cases. Occasional CD31 positivity may occur, however, ES has never been shown to stain for HFVIII.
Cytology specimens usually are hypercellular, and show discohesive or loosely clustered medium-to-large epithelioid or spindled cells. The cytoplasm is well-defined and abundant, with occasional vacuoles and dense globoid paranuclear condensations. The eccentric nucleus shows occasional multinucleation, atypia and nucleoli. Closely associated with the cells is a characteristic hyaline fibrillar eosinophilic material that resembles basement membrane and stains magenta pink with May-Grunwald-Giemsa stain. Background collagen fragments, necrosis, or inflammation may also be observed.
Electron microscopy typically demonstrates striking paranuclear whorls or masses of intermediate filaments, along with prominent Golgi and free ribosomes.
Staining patterns are similar for all variants of epithelioid sarcoma. The dense collagenous extracellular matrix highlights with Alcian blue and hyaluronidase. Trichrome stain yields a strong red hue to the cytoplasm. ES strongly stains with low and high-molecular weight cytokeratins, EMA, and vimentin. They are often positive for CD34 (~50%), nuclear cyclin D1, CD7, and CA-125. They may also be positive for muscle-specific actin and GLUT-1. Unlike other soft tissue sarcomas, ES often demonstrates intratumoral lymphatics by Lyve-1/podoplanin staining. Staining for p63 and cytokeratin 5/6 tends to be negative, as are S-100, neurofilaments, carcinoembryonic antigen, von Willebrand factor, factor VIII, and CD31. Frequently, loss of normal INI1 (SMARCB1) expression is observed.
A specific cell of origin has not yet been identified, although mesenchymal, endothelial, synovial, and fibrohistiocytic origins have been proposed. Genetic mutations in 11q, 18q11, and 22q11 have been occasionally observed; however, recent studies have identified inactivating mutations of the tumor suppressor gene INI1 (SMARCB1) at 22q11 in >90% of epithelioid sarcomas. This mutation can also be seen in some epithelioid malignant peripheral nerve sheath tumors, myoepithelial carcinomas, atypical teratoid/rhabdoid tumors of the central nervous system, and 80% of renal and malignant extrarenal rhabdoid tumors (MERT). The original definition of MERT was not always clearly defined in earlier studies and reports; thus, some adult cases of MERT may overlap with the large cell (‘rhabdoid’, ‘proximal-type’) variant of epithelioid sarcoma. Further studies are needed to establish more precise definitions based on the recent genetic findings.
Despite its usual appearance as a low to medium grade tumor, ES prognosis is very poor. It is exceptionally bad when occurring in axial (“proximalâ€) or deep locations. Increased aggressiveness is also seen in males, a tumor size (>5 cm), increased mitotic rates, and abnormal expression of dysadherin. The presence of hemorrhage, necrosis, vascular invasion, or an inadequate initial excision also portend poor prognosis. There is a high risk of tumor recurrence, often within the first year of diagnosis; however, recurrence may be seen as many as twenty years later. Metastases are frequent and often involve regional lymph nodes or lung. The overall 5 year survival is 50-85%, and the 10 year survival is 40-55%.
Because of ES known proclivity for relentless proximal progression, frequent recurrences, and metastases, the recommended therapy is early, complete excision by amputation or radical local excision. A few studies have reported equivocal results in limb-sparing versus amputation of localized lesions, but these study sizes were small and follow-up was limited. Regional lymph node dissection or at least sentinel lymph node biopsy should be performed, especially if radiographic findings are suspicious. Radiation and chemotherapy may provide some benefit, but cannot replace surgery. Recent chemotherapy have utilized include sunitinib malate, navelbine, anthracycline with ifosfamide, trabectedin, paclitaxel with 5-FU, and erlotinib (EGFR) with rapamycin. Long term follow-up is essential.
Chase and Enzinger’s 1985 paper highlighted a better prognosis in females and recommended investigation into hormone effects on tumor therapy. Recent case reports have described a connection to hormones in certain cases, including human chorionic gonadotropin expression with placenta metastases and the presentation and diagnosis of vulvar epithelioid sarcomas during pregnancy. Further research into hormone therapy may prove useful.
The differential diagnosis of ES includes benign inflammatory processes, epithelioid malignant peripheral nerve sheath tumor (MPNST), melanoma, epithelioid angiosarcoma, epithelioid sarcoma-like hemangioendothelioma, synovial sarcoma, ulcerating squamous cell carcinoma, and malignant extrarenal rhabdoid tumor (MERT).
• Numerous benign inflammatory processes may be considered in the differential for ES, including necrotizing infectious granuloma, necrobiosis lipoidica, granuloma annulare, rheumatoid nodule, nodular fasciitis, fibrous histiocytoma, and fibromatosis. Compared to these processes, the individual cells in epithelioid sarcoma are more sharply defined, larger, and more eosinophilic with prominent epithelioid features and architectural nodularity. The nuclei are more vesicular than are seen in palisading histiocytes. When occurring in the skin, the necrobiotic variant of epithelioid sarcoma mimics granuloma annulare and rheumatoid nodule, but may easily be distinguished by the expression of cytokeratin and by CD34 positivity (~50%). Nodular fasciitis tends to be GLUT-1 negative, while ES may show positivity (~50%).
• ES may grow along nerve bundles, mimicking epithelioid malignant peripheral nerve sheath tumor (MPNST). However, MPNSTs tend to be positive for S100 (approximately 50%) and are uniformly negative for cytokeratins. EMA, however, is only rarely expressed in MPNST.
• Melanoma may also be confused with ES, especially the large cell (“rhabdoidâ€, “proximalâ€) variant. Staining for S100 and other melanocyte markers (HMB-45, Melan A) will likely be positive in melanoma but are negative in ES.
• Epithelioid angiosarcoma may share ES’s loss of cellular cohesion, secondary hemorrhage, and large epithelioid cells with cytoplasmic vacuolation. Although epithelioid angiosarcoma may express cytokeratins and CD34; it strongly stains for endothelial markers such as von Willebrand factor and CD31.
• Epithelioid sarcoma-like hemangioendothelioma (EH) occurs in the extremities of young to middle aged adults and microscopically has vascular lumen formations, which are mimicked by ES’s occasional intracellular lipid droplets. Although EH may also express cytokeratin and vimentin, it marks for vascular markers CD31, HFVIII, and FLI-1, and unlike ES, CD-34 is uniformly negative.
• Biphasic synovial sarcoma shows an abrupt transition between epithelial and spindle patterns, unlike ES’s smoother transition. Synovial sarcoma tends to affect the proximal portion of extremities and is less likely to involve superficial structures and skin. It may share positive immunohistochemical staining for cytokeratin and EMA with ES, but stains negatively for CD34 and shows a characteristic (X;18) translocation.
• Ulcerating squamous cell carcinoma may share a similar clinical presentation as ES; however, it may demonstrate keratin pearls and dyskeratosis in adjacent epithelium, along with strong positivity for CD5/6 and p63. It lacks cyclinD1 (nuclear) and CD34 expression.
• Malignant extrarenal rhabdoid tumor (MERT) in soft tissue should be reserved for tumors with prominent rhabdoid features but lacking a clear line of differentiation. This aggressive tumor typically presents in children as a fleshy, necrotic lesion. It may overlap with the large cell (‘proximal’, ‘rhabdoid’) variant of ES, sharing similar rhabdoid cells, PAS-positive hyaline inclusions of intermediate filament whorls, staining patterns, and INI1 gene mutations (22q11).
Suggested Reading:
1. Weiss SW, Goldblum JR. Enzinger & Weiss’s Soft Tissue Tumors. 5th ed. Philadelphia: Mosby-Elsevier, 2008:1191-1203, 1208-1213.
2. Chase DR. Do “rhabdoid features†impart a poorer prognosis to proximal-type epithelioid sarcomas? Adv Anat Pathol. 1997;4(5):293-299. Regarding: Guillou L, Wadden C, Coindre JM, Krauz T, Fletcher CDM. ‘Proximal-type’ epithelioid sarcoma, a distinctive aggressive neoplasm showing rhabdoid features: clinicopathologic, immunohistochemical, and ultrastructural study of a series. Am J Surg Pathol. 197;21(2):130-146.
3. Chase DR, Enzinger FM. Epithelioid sarcoma: diagnosis, prognostic indicators, and treatment. Am J Surg Pathol. 1985;9(4):241-263.
4. Enzinger FM. Epithelioid sarcoma: a sarcoma simulating a granuloma or a carcinoma. Cancer. 1970;26:1029-1041.
5. Chase DR. Letters to the editor: rhabdoid versus epithelioid sarcoma. Am J Surg Pathol. 1990;14(8):792-793. Regarding: Perrone T, Swanson PE, Twiggs L, et al. Malignant rhabdoid tumor of the vulva: is distinction from epithelioid sarcoma possible? Am J Surg Pathol. 1989;13:848-858.
6. Chase DR. Epithelioid sarcoma. California Tumor Tissue Registry. Berlin, Germany. September 12, 2001. Seminar Presentation of Epithelioid Neoplasms.
7. Sur M, Nayler SJ. Proximal epithelioid sarcoma – a misnomer. Histopathology. 2001;39(6):641-643.
8. Rakheja D, Wilson KS, Meehan J, Schultz RA, Gomez AM. “Proximal-type†and classic epithelioid sarcomas represent a clinicopathologic continuum: case report. Pediatr Dev Pathol. 2005;8(1):105-114.
9. Boutilier R, Walsh N. Pathologic quiz case: cutaneous nodule of 3-year duration: Epithelioid sarcoma. Arch Pathol Lab Med. 2002;126(5):625-626.
10. Gupta H, Davidoff AM, Rao BN, Jenkins JJ, Spunt SL. Neonatal epithelioid sarcoma: a distinct clinical entity? J Pediatr Surg. 2006;41(7):e9-e11.
11. Keelawat S, Shuangshoti S, Kittikowit W, Lerdlum S, Thorner PS. Epithelioid sarcoma of the parotid gland of a child. Pediatr Dev Pathol. 2009;12(4):301-306.
12. Bento A, Baptista H, Pinheiro C, Martinho F. Jejuno-jejunal invagination caused by epithelioid sarcoma: a case report. J Med Case Rep. 2009;3:89.
13. Maggiani F, Debiec-Rychter M, Ectors N, Lerut A, Sciot R. Primary epithelioid sarcoma of the oesophagus. Virchows Arch. 2007;451(4):835-838.
14. Raoux D, Peoc’h M, Pedeutour F, Vaunois B, Decouvelaere AV, Folpe AL. Primary epithelioid sarcoma of bone: report of a unique case, with immunohistochemical and fluorescent in situ hybridization confirmation of INI1 deletion. Am J Surg Pathol. 2009;33(6):954-958.
15. Kosemehmetoglu K, Kaygusuz G, Bahrami A, Raimondi SC, Kilicarslan K, Yildiz Y, Folpe AL. Intra-articular epithelioid sarcoma showing mixed classic and proximal-type features: report of 2 cases with immunohistochemical and molecular cytogenetic INI-1 study. Am J Surg Pathol. 2011;35(6):891-897.
16. Choi SY, Kim YH, Kwon JB, Suh JH, Shin OR, Hong SH. Epithelioid sarcoma metastatic to the lung as pulmonary cysts without other metastatic manifestation. J Thorac Oncol. 2008;3(5):532-533.
17. Barnoud R, Collard-Frachon S, de la Roche E, Vasiljevic A, Thomson V, Ranchere D, Devouassoux G, Devouassoux-Shisheborn M. Lung metastases of epithelioid sarcoma revealed by bilateral spontaneous pneumothorax: a pathological diagnosis. Ann Pathol. 2010;30(2):139-142.
18. Jashnani KD, Dhume VM, Bahal NK. Epithelioid sarcoma and squamous cell carcinoma arising in a burn scar. Indian J Dermatol. 2011;56(5):585-586.
19. Kagami S, Saeki H, Idezuki T, Yano S, Kawabata Y, Okochi H, Asahina A, Nakagawa K, Tamaki K. Epithelioid sarcoma associated with lung adenocarcinoma. J Dermatol. 2005;32(11):904-908.
20. Kimball Dalton VM, Gelber RD, Li F, Donnelly MJ, Tarbell NJ, Sallan SE. Second malignancies in patients treated for childhood acute lymphoblastic leukemia. J Clin Oncol. 1998; 16(8):2848-2853.
21. Rose DS, Fisher C, Smith ME. Epithelioid sarcoma arising in a patient with neurofibromatosis type 2. Histopathology. 1994;25(4):379-380.
22. Kierson ME, Iyer D, Fyfe B. Metastatic epithelioid sarcoma in a non-healing ventricular device pocket. J Heart Lung Transplant. 2010;29(11):1319.
23. Hoshino M, Kawashima H, Ogose A, Kudo N, Ariizumi T, Hotta T. Serum CA-125 expression as a tumor marker for diagnosis and monitoring the clinical course of epithelioid sarcoma. J Cancer Res Clin Oncol. 2010;136(3):457-464.
24. Sakamoto A, Jono O, Hirahashi M, Oya M, Iwamoto Y, Arai K. Epithelioid sarcoma with muscle metastasis detected by positron emission tomography. World J Surg Oncol. 2008;6:84.
25. Dion E, Forest M, Brasseur JL, Amoura Z, Grenier P. Epithelioid sarcoma mimicking abscess: review of the MRI appearances. Skeletal Radiol. 2001;30(3):173-177.
26. Kozawa E, Irisawa M, Heshiki A, Okagaki R, Shimizu Y. Magnetic resonance imaging findings of vulvar epithelioid sarcoma. Radiat Med. 2008;26(6):376-378.
27. Barwad A, Dey P, Das A. Fine needle aspiration cytology of epithelioid sarcoma. Diagn Cytopathol. 2010;39(7):517-520.
28. Lemos MM, Chaves P, Mendonca ME. Is preoperative cytologic diagnosis of epithelioid sarcoma possible? Diagn Cytopathol. 2008;36(11):780-786.
29. Laskin WB, Miettinen M. Epithelioid sarcoma: new insights based on an extended immunohistochemical analysis. Arch Pathol Lab Med. 2003;127(9):1161-1168.
30. Miettinen M, Fanburg-Smith JC, Virolainen M, Shmookler BM, Fetsch JF. Epithelioid sarcoma: an immunohistochemical analysis of 112 classical and variant cases and a discussion of the differential diagnosis. Hum Pathol. 1999;30(8):934-942.
31. Izumi T, Oda Y, Hasegawa T, Nakanishi Y, Iwasaki H, Sonobe H, et al. Prognostic significance of dysadherin expression in epithelioid sarcoma and its diagnostic utility in distinguishing epithelioid sarcoma from malignant rhabdoid tumor. Mod Pathol. 2006;19(6):820-831.
32. Chu PG, Arber DA, Weiss LM. Expression of T/NK-cell and plasma cell antigens in nonhematopoietic epithelioid neoplasms: an immunohistochemical study of 447 cases. Am J Clin Pathol. 2003;120(1):64-70.
33. Lin L, Hicks D, Xu B, Sigel JE, Bergfeld WF, Montgomery E, et al. Expression profile and molecular genetic regulation of cyclin D1 expression in epithelioid sarcoma. Mod Pathol. 2005;18(5):705-709.
34. Mahendra G, Kliskey K, Williams K, Hollowood K, Jackson D, Athanasou NA. Intratumoral lymphatics in benign and malignant soft tissue tumours. Virchows Arch. 2008;453(5):457-464.
35. Orrock JM, Abbott JJ, Gibson LE, Folpe AL. INI1 and GLUT-1 expression in epithelioid sarcoma and its cutaneous neoplastic and nonneoplastic mimics. Am J Dermatopathol. 2009;31(2):152-156.
36. Hornick JL, Dal Cin P, Fletcher CD. Loss of INI1 expression is characteristic of both conventional and proximal-type epithelioid sarcoma. Am J Surg Pathol. 2009;33(4):542-550.
37. Kohashi K, Izumi T, Oda Y, Yamamoto H, Tamiya S, Taguchi T. Infrequent SMARCB1/INI1 gene alteration in epithelioid sarcoma: a useful tool in distinguishing epithelioid sarcoma from malignant rhabdoid tumor. Human Pathol. 2009;40(2):349-355.
38. Argenta PA, Thomas S, Chura JC. Proximal-type epithelioid sarcoma vs. malignant rhabdoid tumor of the vulva: a case report, review of the literature, and an argument for consolidation. Gynecol Oncol. 2007;107(1):130-135.
39. Guzzetta AA, Montgomery EA, Lyu H, Hooker CM, Meyer CF, Loeb DM, et al. Epithelioid sarcoma: one institution’s experience with a rare sarcoma. J Surg Res. 2012;177(1):116-122.
40. Gasparini P, Facchinetti F, Boeri M, Lorenzetto E, Livio A, Gronchi A, et al. Prognostic determinants in epithelioid sarcoma. Eur J Cancer. 2011;47(1):287-295.
41. Sakharpe A, Lahat G, Gulamhusein T, Liu P, Bolshavkov S, Nguyen T, et al. Epithelioid sarcoma and unclassifiable sarcoma with epithelioid features: clinicopathological variables, molecular markers, and a new experimental model. Oncologist. 2011;16(4):512-522. .
42. Jawad MU, Extein J, Min ES, Scully SP. Prognostic factors for survival in patients with epithelioid sarcoma: 441 cases from the SEER database. Clin Orthop Relat Res. 2009;467(11):2939-2948.
43. Jones RL, Constantinidou A, Olmos D, Thway K, Fisher C, Al-Muderis O, Scurr M, Judson IR. Role of palliative chemotherapy in advanced epithelioid sarcoma. Am J Clin Oncol. 2012;35(4):351-357.
44. Xie X, Ghadimi MP, Young ED, Belousov R, Zhu QS, Liu J, Lopez G, Colombo C, Peng T, Reynoso D, Hornick JL, Lazar AJ, Lev D. Combining EGFR and mTOR blockage for the treatment of epithelioid sarcoma. Clin Cancer Res. 2011;17(18):5901-5912.
45. De Visscher SA, van Ginkel RJ, Wobbes T, Veth RP, Ten Heuvel SE, Suurmeijer AJ, Hoekstra HJ. Epithelioid sarcoma: still an only surgically curable disease. Cancer. 2006;107(3):606-612.
46. Steffensen TS, Gilbert-Barness E, Wagoner MJ, Bui MM, Browarsky IL. Human chorionic gonadotropin producing epithelioid sarcoma metastatic to the placenta. Fetal Pediatr Pathol. 2008;27(6):282-291.
47. Rai H, Odunsi K, Kesterson J, et al. Epithelioid sarcoma of the vulva in a 17-year-old pregnant woman. Appl Immunohistochem Mol Morphol. 2009;17(3):270-273.
48. Moore RG, Steinhoff MM, Granai CO, DeMars LR. Vulvar epithelioid sarcoma in pregnancy. Gynecol Oncol. 2002;85(1):218-222.