History: A 14 month old girl presented with hepatomegaly, with the palpable liver margin 5 cm below the inferior mid-clavicular line. Shortly after presenting, the patient died, and an autopsy described an enlarged liver weighing 1015 grams. Sectioning showed a yellow-green, 8 x 4 cm variegated mass with cystic, gelatinous, and focally calcified areas, located anteriorly between the left and right lobes.
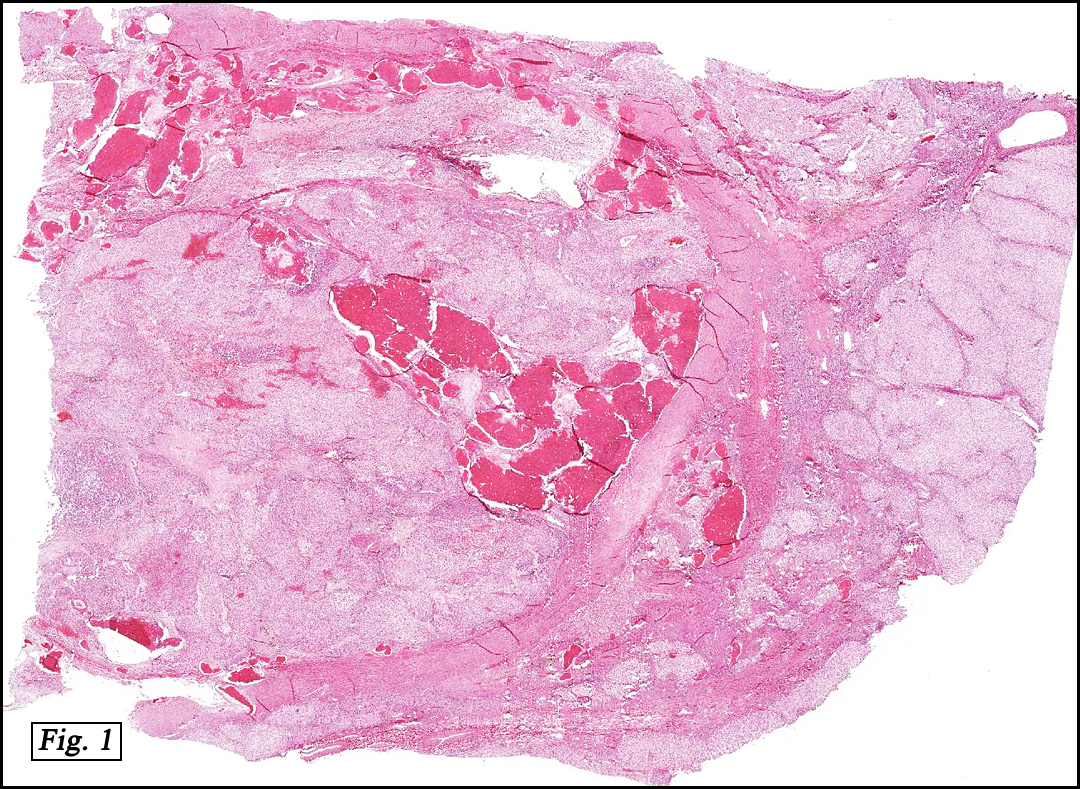
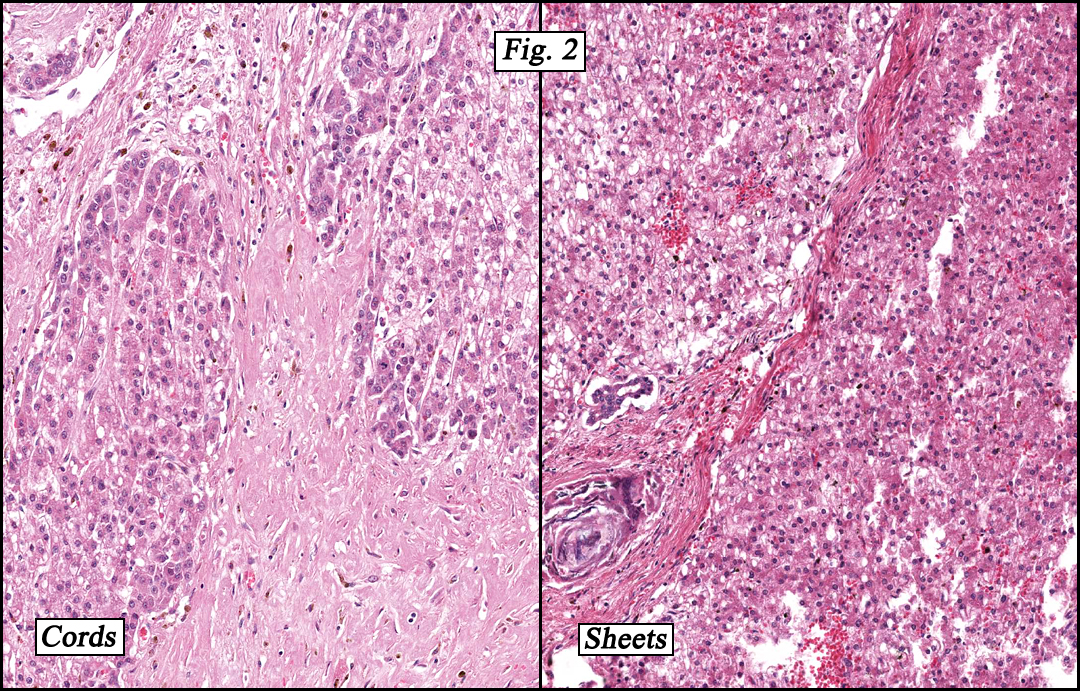
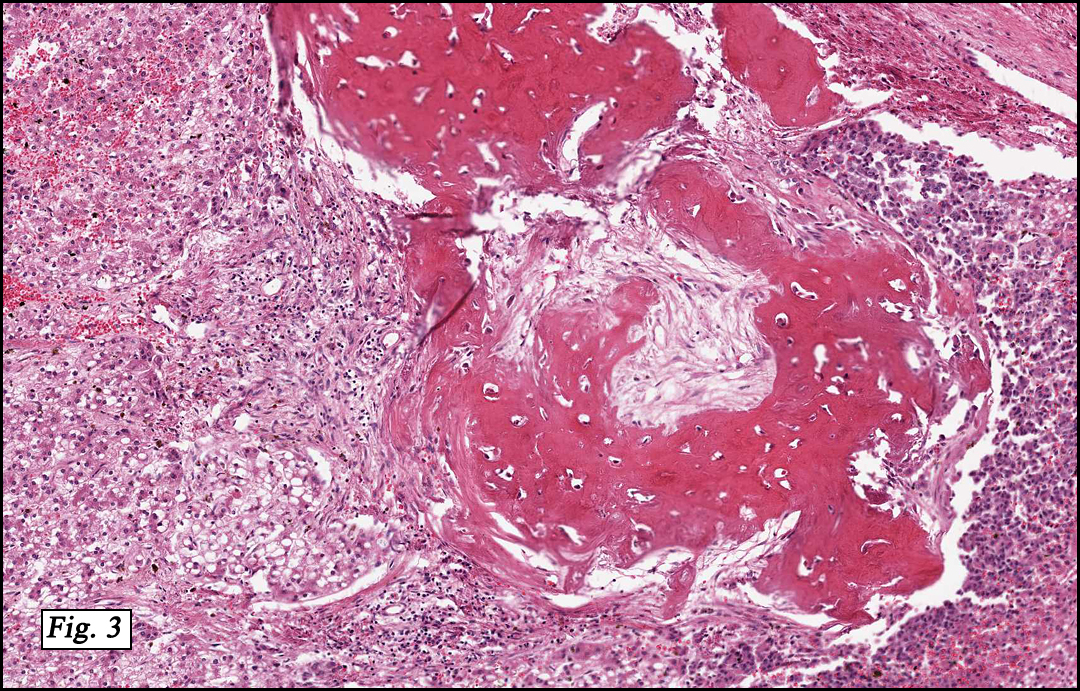
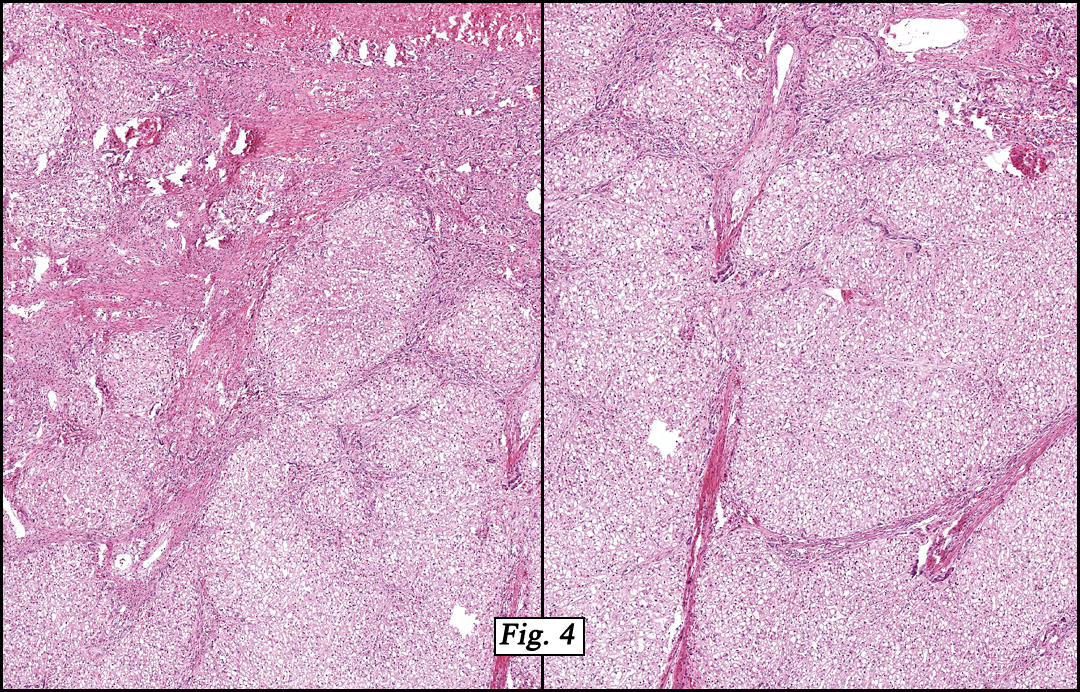
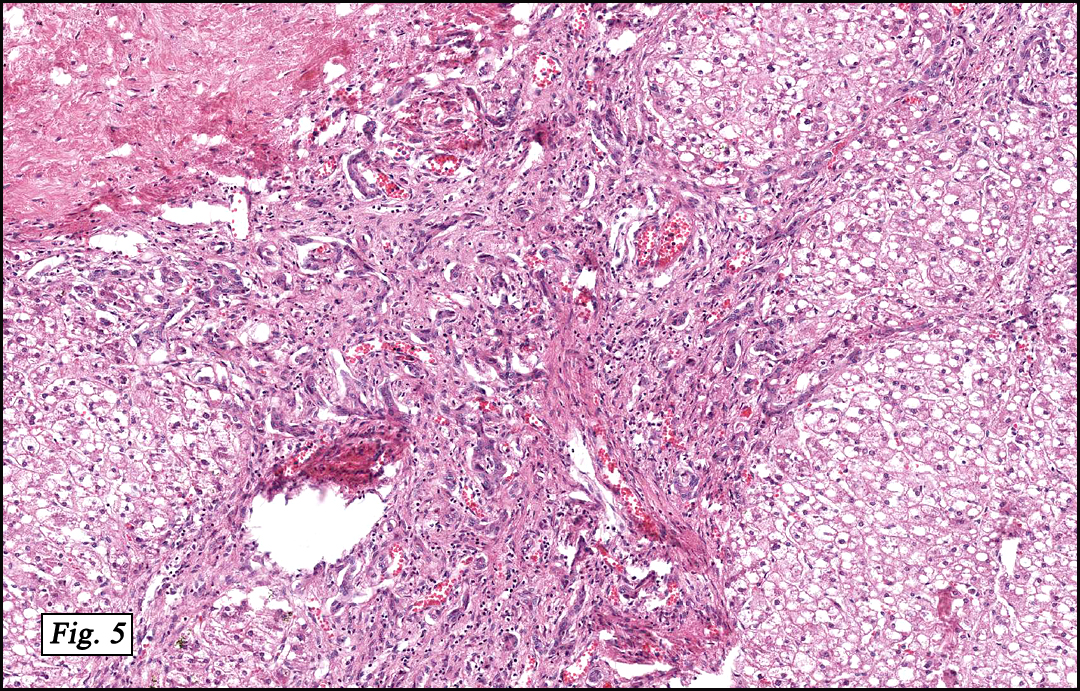
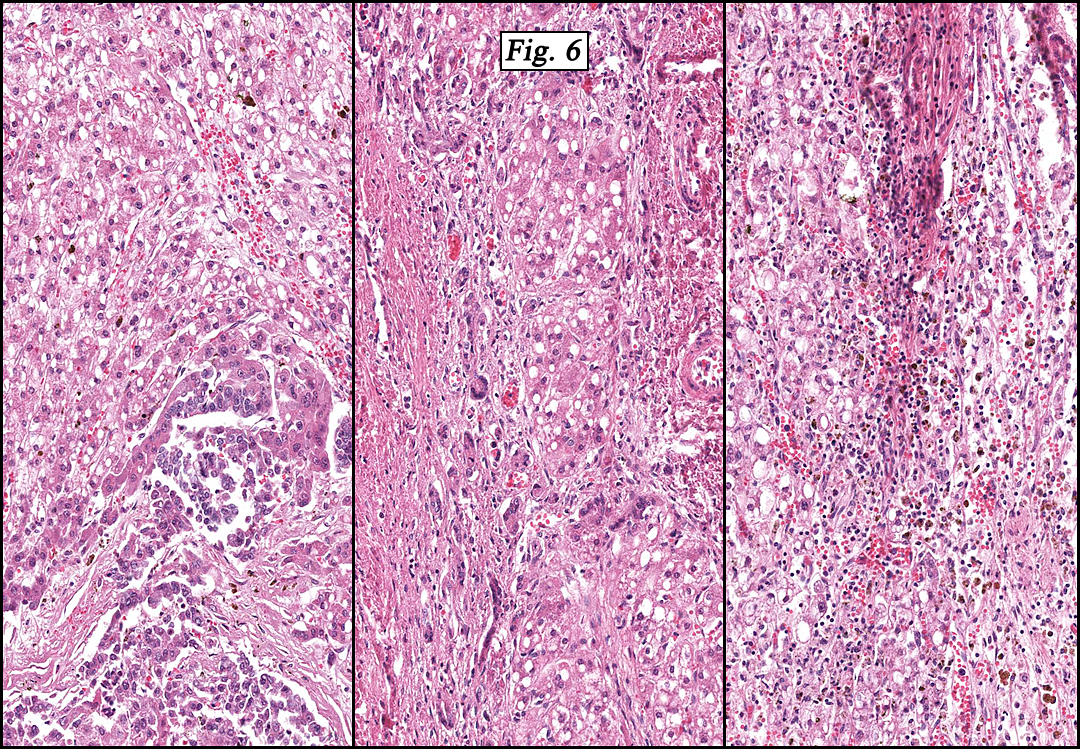
Microscopically, the tumor was partially encapsulated, lobulated, and exhibited patchy light and dark regions. Some areas were telangiectatic with numerous expanded blood vessels filled with erythrocytes (Fig. 1). Growth was usually as cords and sheets (Fig. 2). Abundant osteoid could be seen adjacent to fibrous septa and loose mesenchymal elements (Fig. 3). The lighter staining regions consisted of more mature hepatocytes growing in a lobular pattern separated by fibrous septa (Fig. 4). Proliferation of bile ducts could also be seen (Fig. 5). Also present were clusters of blastemal cells with an increased nuclear to cytoplasm ratio with greater nuclear pleomorphism and prominent nucleoli (Fig. 6, left). Rare multinucleated tumor giant cells were present (Fig. 6, center), and some regions were suggestive of extramedullary hematopoiesis (Fig. 6, right). There were significant areas of hemorrhage and hemosiderin deposition. Mitoses were rare.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Diagnosis: “Hepatoblastoma, Mixed Patternâ€
Shino Magaki MSIV, Anwar Raza MBBS, and Donald R. Chase MD
Department of Pathology and Human Anatomy,
Loma Linda University and Medical Center
California Tumor Tissue Registry, Loma Linda, California
Discussion: Hepatoblastoma (HB) is the most common liver malignancy in children, accounting for approximately two-thirds of all primary pediatric hepatic malignancies and 1% of all pediatric malignancies. Two-thirds are diagnosed prior to the age of 2 years while 90% are diagnosed before the age of 4 years, with the mean age at diagnosis of 18 months. HB is rarely seen at birth or in young adults, and males are twice as likely as females to be affected. HB was probably first described by Misick in 1898. The etiology is unknown but 5% of cases are associated with a congenital abnormality such as Beckwith-Wiedemann syndrome or familial adenomatous polyposis. Common cytogenetic alterations include trisomy 2, 8 and 20. Activating mutations in the beta-catenin/Wnt pathway is seen in about half of the cases. HB has also been associated with low birth weight and has been increasing in incidence over the past few decades, possibly due to improved survival of low birth weight infants.
Clinically, patients often present with a rapidly enlarging abdominal mass, failure to thrive, and weight loss. Less common symptoms include vomiting, diarrhea, and jaundice. Virilization and precocious puberty from human chorionic gonadotropin secreted by the tumor have been reported. Serum alpha-fetoprotein (AFP) is elevated in 90% of cases and is a useful marker for tumor recurrence. Tumors that do not express AFP are thought to be more biologically aggressive, and serum levels less than 100 ng/mL at the time of diagnosis indicate a poorer prognosis. On ultrasound, HB usually appears as a well demarcated solitary mass although it can be multifocal. CT often shows contrast enhancement and calcification. On MRI the purely epithelial subtype appears homogenous and hypointense on T1 and hyperintense on T2 weighted images, whereas the mixed epithelial and mesenchymal subtype is more heterogeneous.
Grossly, HB typically occurs as a single mass on a background of non-cirrhotic liver and may contain areas of hemorrhage and necrosis. The tumor tends to be well circumscribed but unencapsulated or partially encapsulated and have a variegated appearance depending on the proportion of its components. In pure epithelial HB, the tumor is soft, fleshy and tan-white. Mixed tumors show a heterogeneous surface, tend to be firmer and often are calcified. Frequently large at detection, HB can measure up to 20-25 cm in diameter (average 10-12 cm), and usually weigh over 1 kg. They are most common in the right lobe.
Histologically, HBs are classified into pure epithelial or mixed epithelial-mesenchymal subtypes with six main histological patterns:
The fetal pattern (30%) demonstrates tumor cells which are uniform and similar in size to normal hepatocytes with a slightly increased nuclear:cytoplasm (N:C) ratio. Cells form trabeculae 2-3 cells thick that are separated by sinusoids. An alternating light and dark staining pattern due to lipid and glycogen, respectively, is often seen. Portal tracts and bile ducts and ductules are absent, and mitoses are rare (
In the embryonal pattern (20%), cells are less differentiated, have a higher N:C ratio with coarser chromatin, and may form sheets. More mitoses and necrosis are seen.
Cells are the least differentiated in the anaplastic small cell pattern (3%), forming sheets of small cells with scant cytoplasm and prominent nucleoli, resembling small blue cell tumors elsewhere. Tumor cells are positive for CK and negative for bile.
In the macrotrabecular pattern (3%), fetal or embryonal cells form trabeculae 10 or more cells thick throughout the tumor.
The mixed epithelial-mesenchymal pattern (44%) is characterized by the presence of both epithelial and mesenchymal cells. The mesenchymal component may show primitive stellate-shaped cells with little cytoplasm or demonstrate chondroid and rhabdomyoblastic differentiation. Osteoid is the most common heterologous element and is more frequent found after chemotherapy. The mixed pattern can be further subdivided into tumors with (10%) or without (34%) teratoid features such as skeletal muscle, keratinized squamous epithelium, and intestinal epithelium.
Differential Diagnoses:
Although Hepatocellular carcinoma (HCC) may be indistinguishable from a macrotrabecular pattern of HB, other features help differentiate them (Table 1).
Since small cell tumors, including neuroblastoma, rhabdomyosarcoma, and lymphoma, resemble the anaplastic small cell pattern in HB, the presence of more typical, better differentiated foci should be looked for. Immunohistochemistry is useful, with HB showing CK positivity and neurofilament, desmin, leukocyte common antigen (CD45) negativity; neuroblastoma is positive for neurofilaments, rhabdomyosarcoma is positive for desmin, and lymphoma is CD45 positive.
Table 1. Hepatoblastoma and hepatocellular carcinoma in the pediatric age group (
| Hepatoblastoma | Hepatocellular carcinoma | |
| Incidence | Most common primary liver malignancy in children (0.5-1.5 diagnoses per million annually) |
2nd most common primary liver malignancy in children |
| Age | 90%<5 yrs | Most >10 years |
| M:F ratio | 1.2-3.6:1 | 1.7:11:1 |
| Association with genetic syndromes |
Familial adenomatous polyposis, Beckwith-Wiedemann syndrome, trisomy 18 |
Glycogen storage disease type IA, hereditary tyrosinemia, Fanconi syndrome |
| Other associations | Parental exposure to environmental toxins, low birth weight |
Cirrhosis, viral hepatitis |
| Serum AFP elevations | 90%, usually highly elevated | 60-80% |
| Single mass | 70-80% | 15-40% |
| Trabeculae | Usually 2-3 cells thick with occasional macrotrabeculae |
Usually>2-3 cells thick |
| Mitoses | Rare | Common |
| Tumor giant cells | Rare | Common |
| Cytoplasmic inclusions (e.g. Mallory’s hyaline, alpha-1-anti-trypsin) |
Absent | Common |
| Extramedullary hematopoiesis |
Common | Absent |
Treatment consists of complete surgical resection with or without neoadjuvant chemotherapy to decrease tumor size prior to surgery. Prognosis is dependent upon the extent of complete resection. At present, the 5-year survival rate is 75%. Liver transplantation is considered when the tumor is limited to the liver but unresectable. The pure fetal pattern is thought to predict a more favorable outcome while the pure anaplastic small cell pattern, increased mitoses, and vascular invasion suggest a poorer prognosis. Most frequent metastatic sites are the regional lymph nodes and lungs.
Suggested Reading:
Beach R, Betts P, Radford M, Millward-Sadler H. Production of human chorionic gonadotrophin by a hepatoblastoma resulting in precocious puberty. J Clin Pathol. 37:734-37, 1984.
Buccoliero AM, Castiglione F, Maio V, Moncini D, Sardi I, Taddei A, Martin A, Messineo A, Taddei GL. Teratoid hepatoblastoma. Fetal Pediatr Pathol. 27:274-81, 2008.
Das CJ, Dhingra S, Gupta AK, Iyer V, Agarwala S. Imaging of paediatric liver tumours with pathological correlation. Clin Radiol. 64:1015-25, 2009.
Litten JB, Tomlinson GE. Liver tumors in children. Oncologist. 13:812-20, 2008.
Maruyama K, Ikeda H, Koizumi T. Hepatoblastoma associated with trisomy 18 syndrome: a case report and a review of the literature. Pediatr Int. 43:302-5, 2001.
Mills SE, Carter D, Greenson JK, Oberman HA, Reuter V, Stoler MH. Sternberg’s Diagnostic Surgical Pathology (4th ed) Philadelphia: Lippincott. 1743-47, 2004.
Reynolds P, Urayama KY, Von Behren J, Feusner J. Birth characteristics and hepatoblastoma risk in young children. Cancer. 100:1070–76, 2004.
Sallam A, Paes B, Bourgeois J. Neonatal hepatoblastoma: two cases posing a diagnostic dilemma, with a review of the literature. Am J Perinatol. 22:413-19, 2005.
Schnater JM, Köhler SE, Lamers WH, von Schweinitz D, Aronson DC. Where do we stand with hepatoblastoma? A review. Cancer. 98:668-78, 2003.