History: A 45-year-old gravida 0 woman presented with frequent menometrorrhagia. Transabdominal sonography and computerized tomography revealed a large multi-loculated cystic mass in the left ovary, multiple nodules in the myometrium, and pelvic lymphadenopathy. The patient was treated with total abdominal hysterectomy, bilateral salphingo-oophorectomy, and limited pelvic lymph node dissection.
The excised uterus and adnexa weighed 2450 grams and contained a multicystic 34.5 x 21.0 x 9.5 cm left ovarian mass dominated by thin and thick-walled cysts filled with mucoid material. The outer surface was smooth. The uterus was 8.0 × 4.5 × 2.5 cm and had unremarkable endometrium and multiple intramural leiomyomata up to 0.8 cm. Both oviducts were normal as was the right ovary. Multiple enlarged pelvic lymph nodes up to 1.5 cm in diameter were present having grey and pink homogeneous cut surfaces. The omentum was unremarkable.
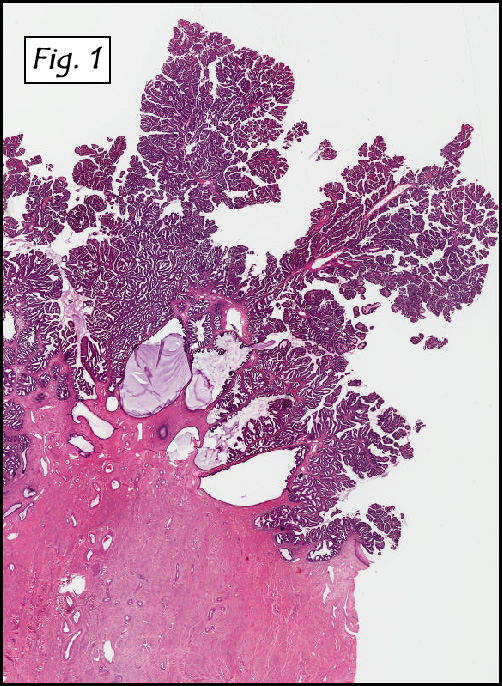
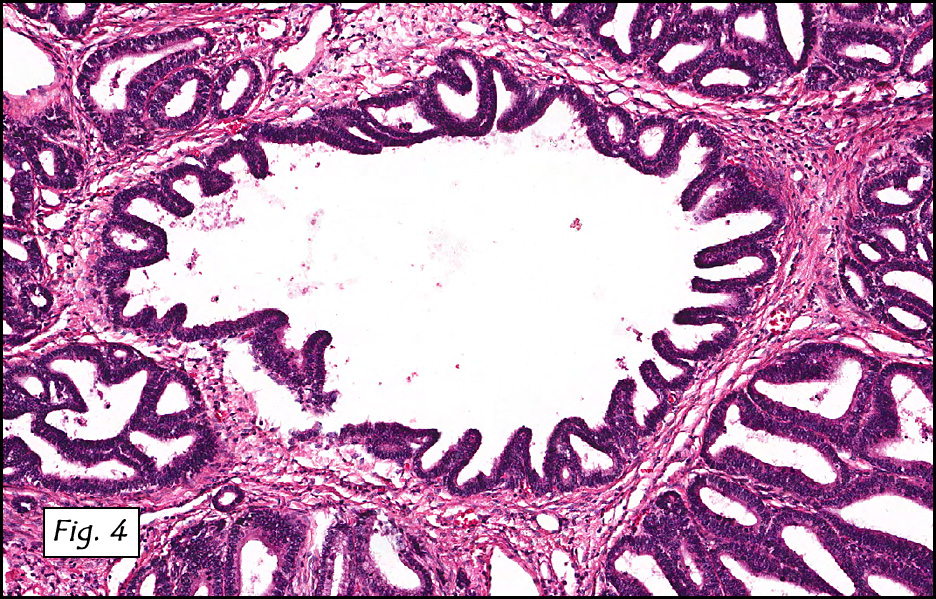
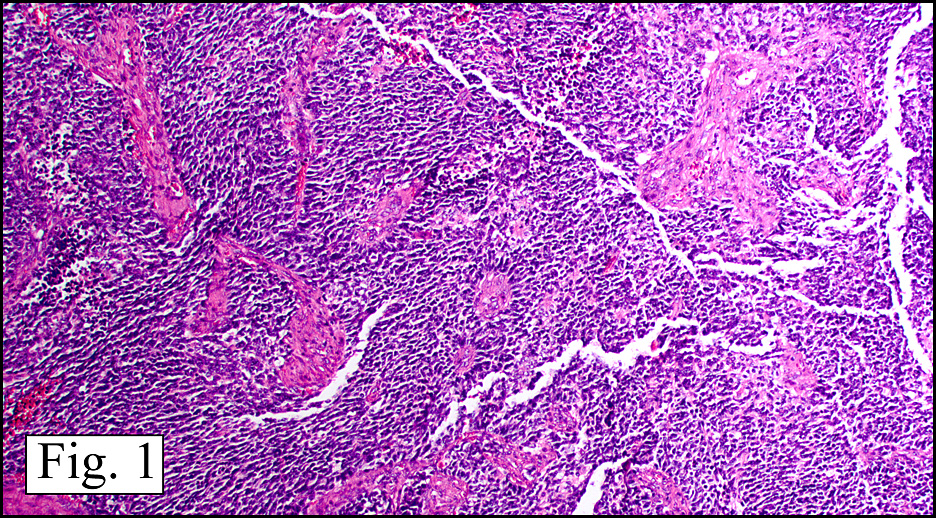
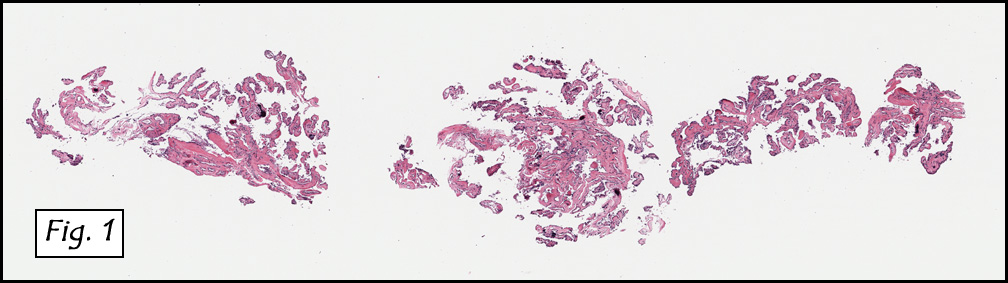
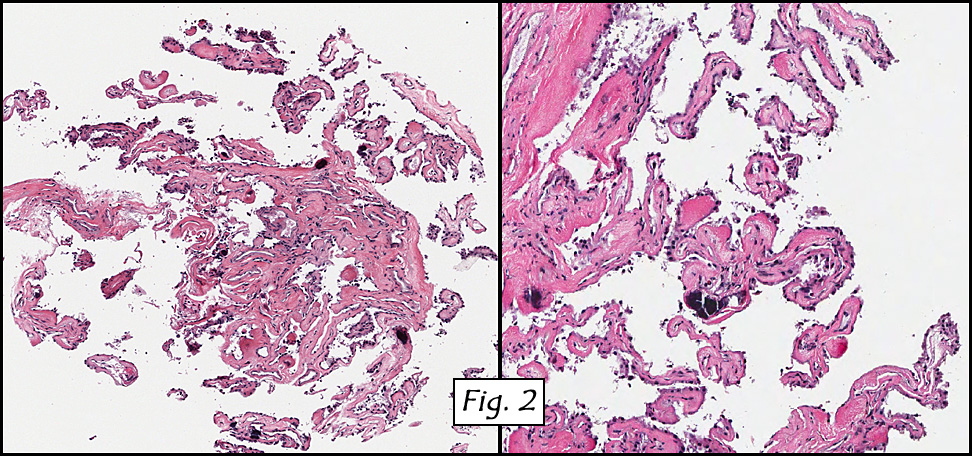
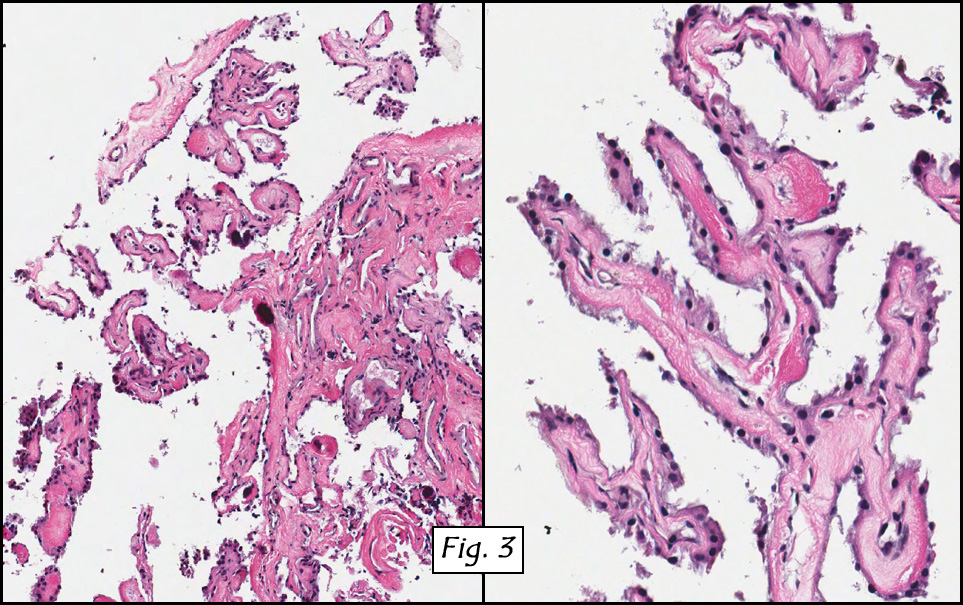
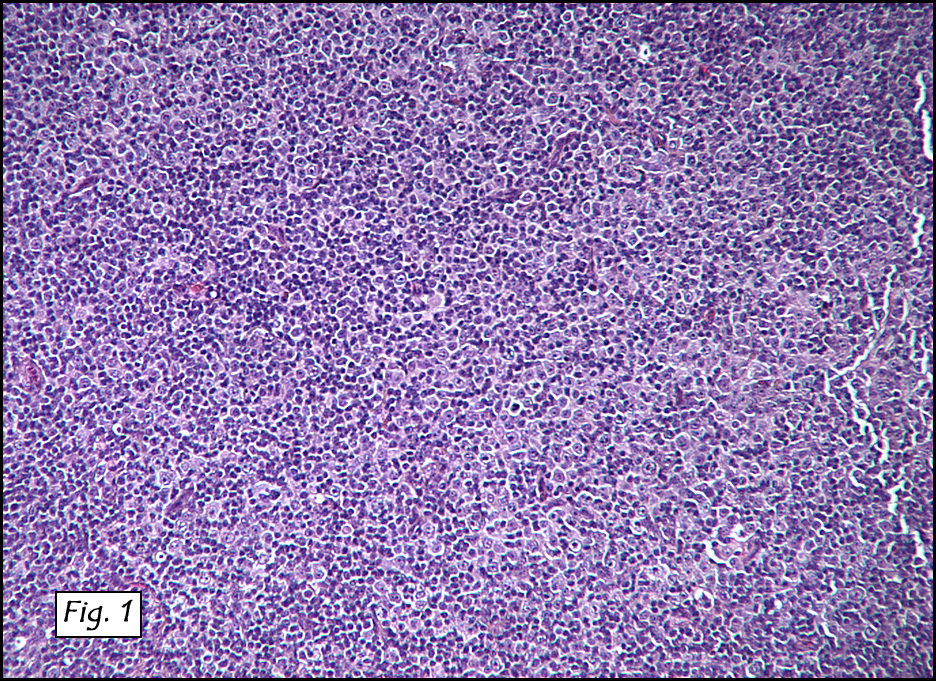
Microscopically, the dominant left ovarian mass had cystic spaces lined by mucin-secreting columnar calls (Fig. 1). Minimal stratification was seen and neither complex microvilli nor marked nuclear pleomorphism were encountered. Stromal invasion was not seen.
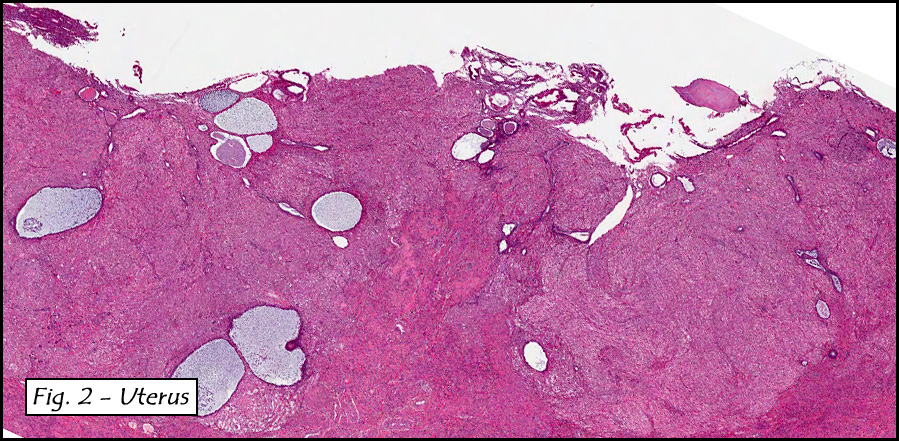
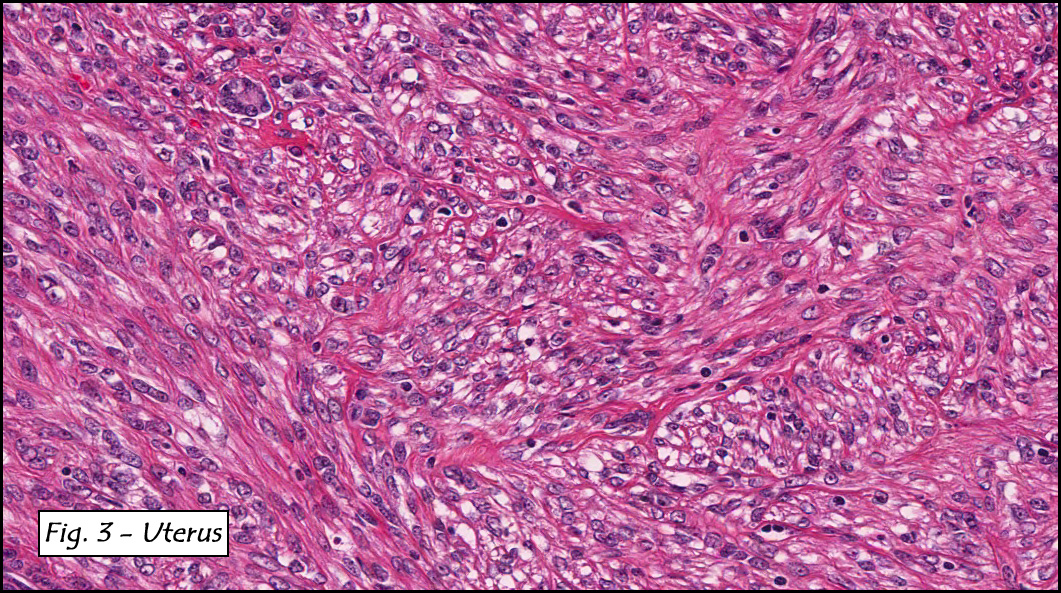
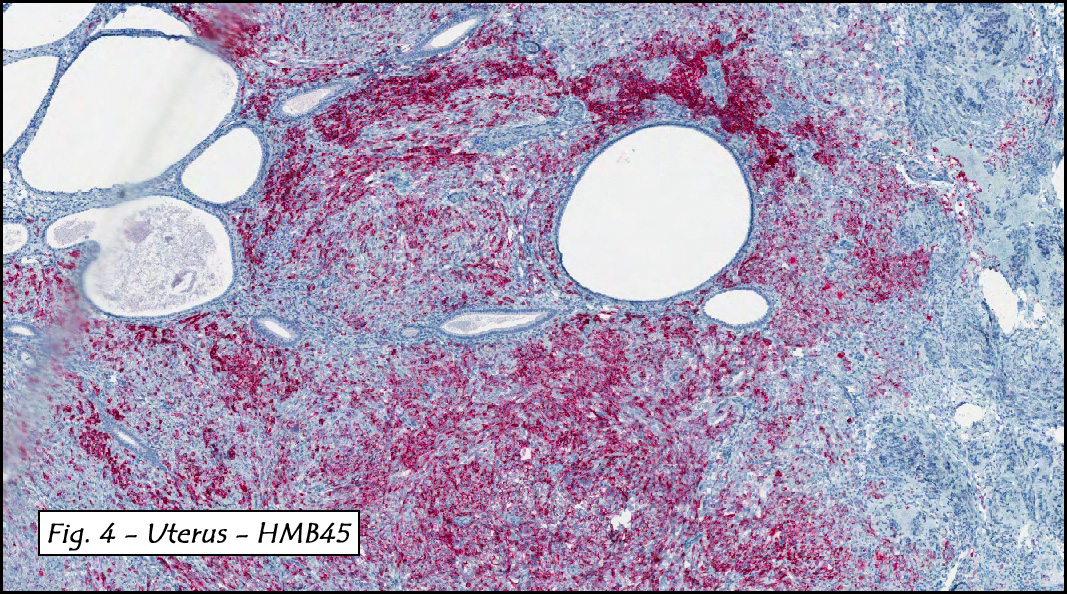
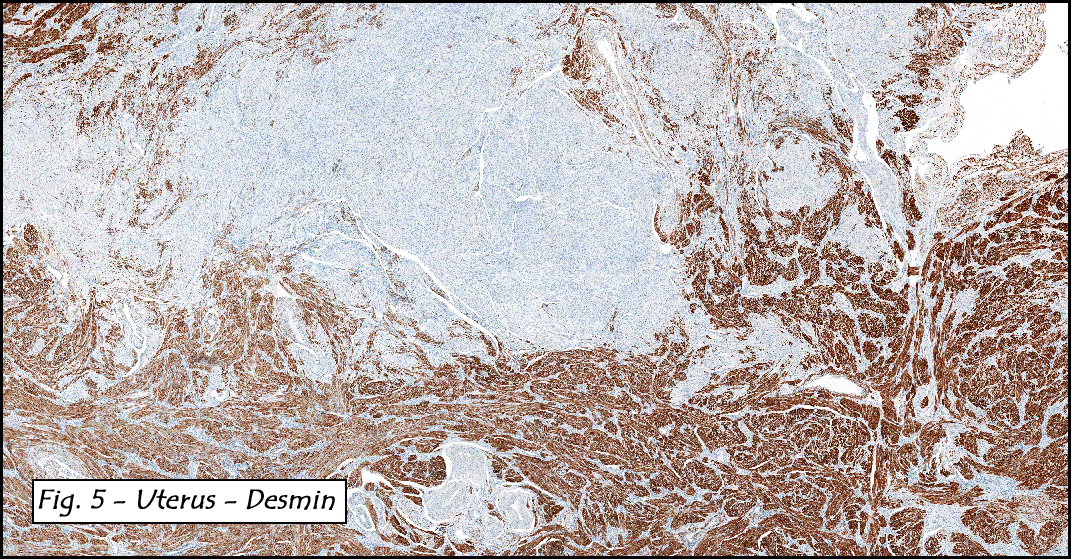
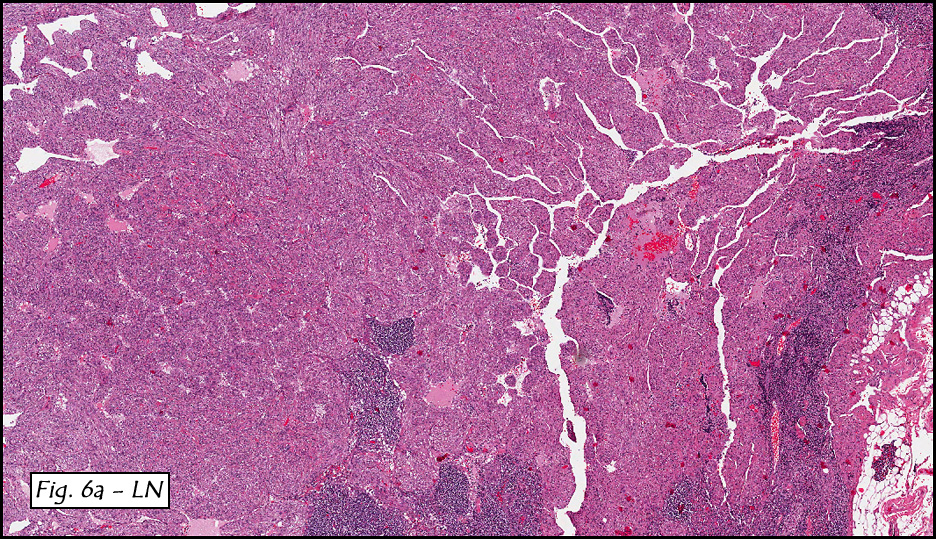
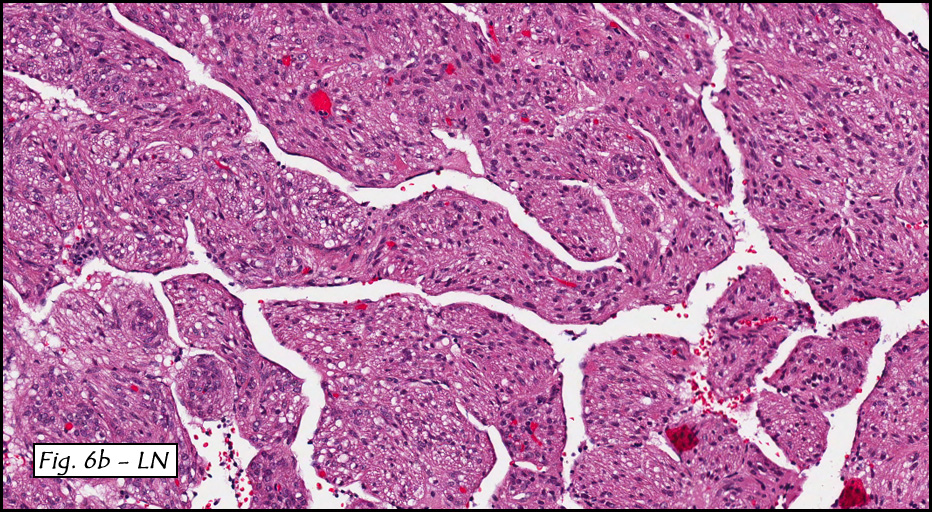
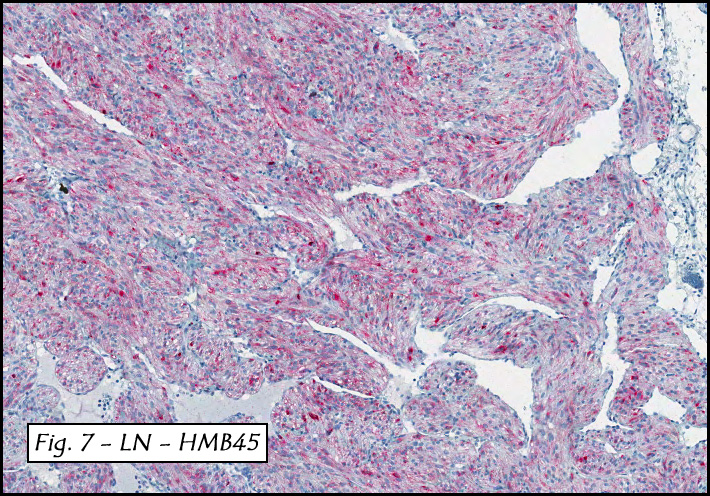
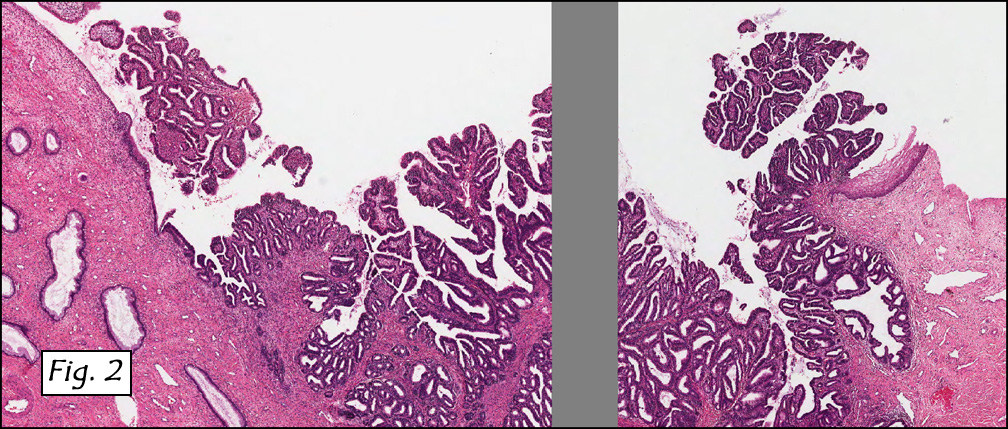
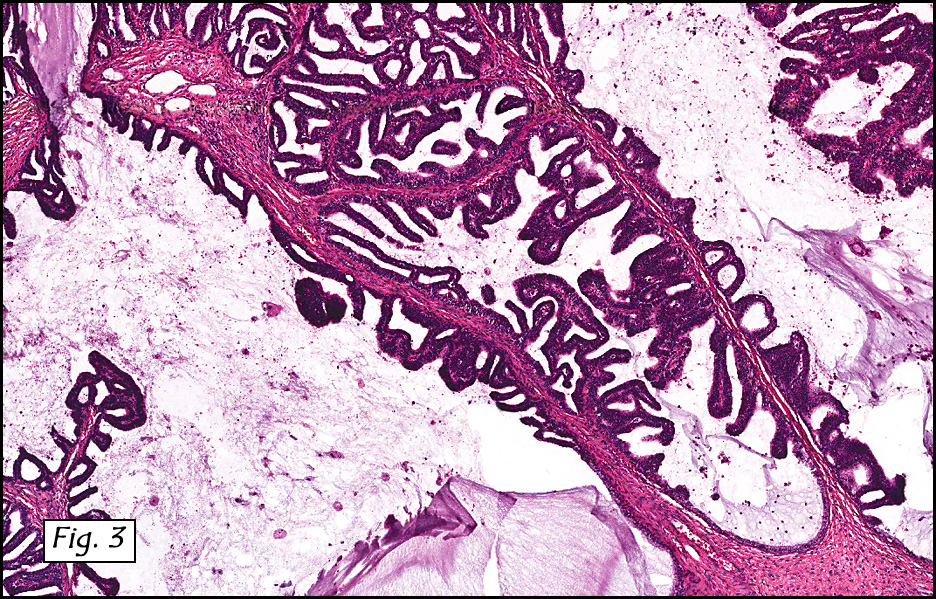
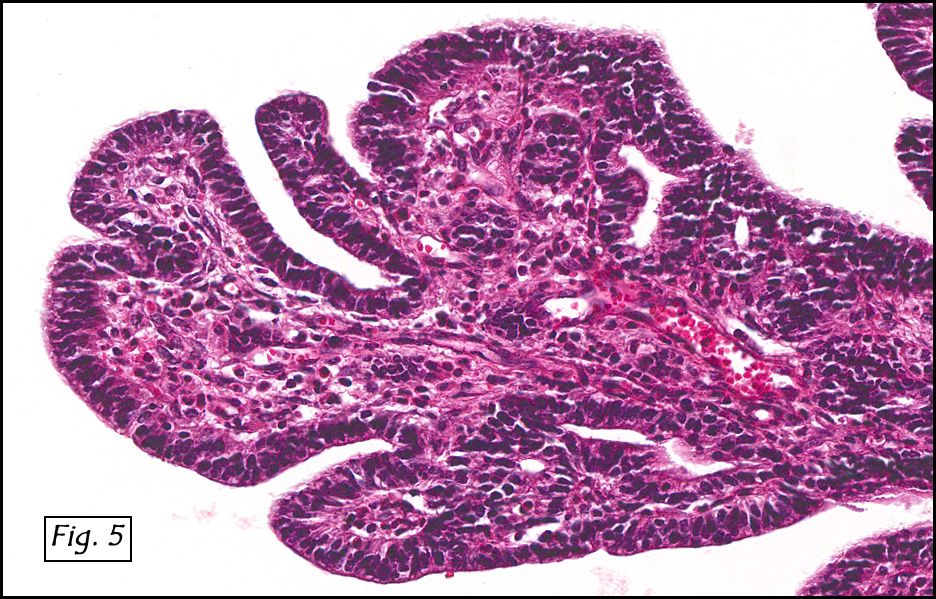
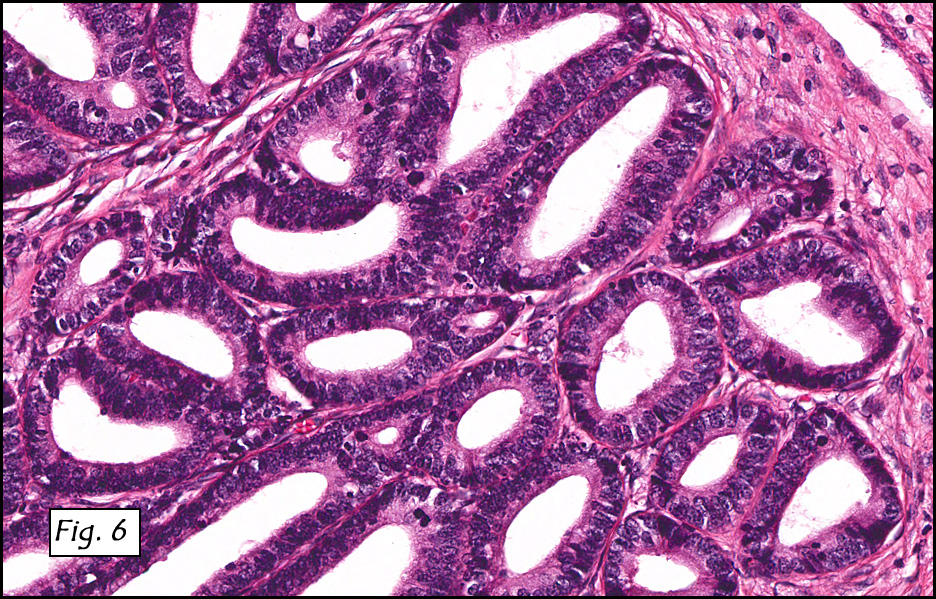
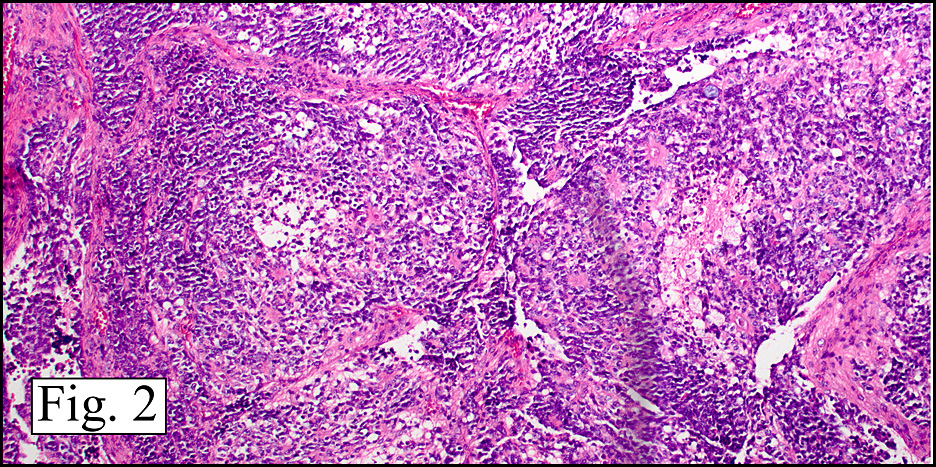
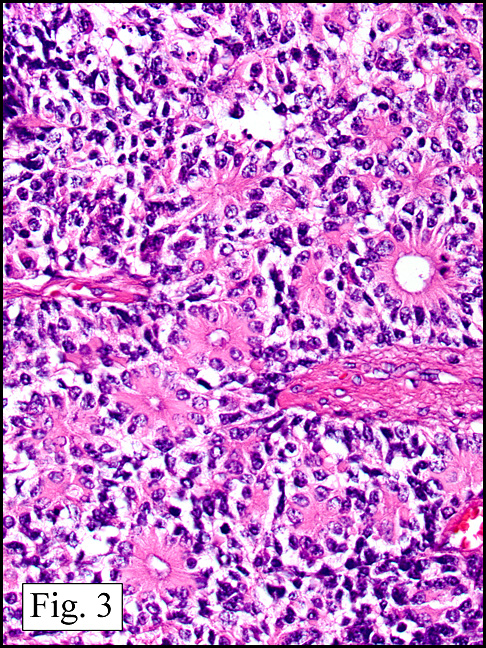
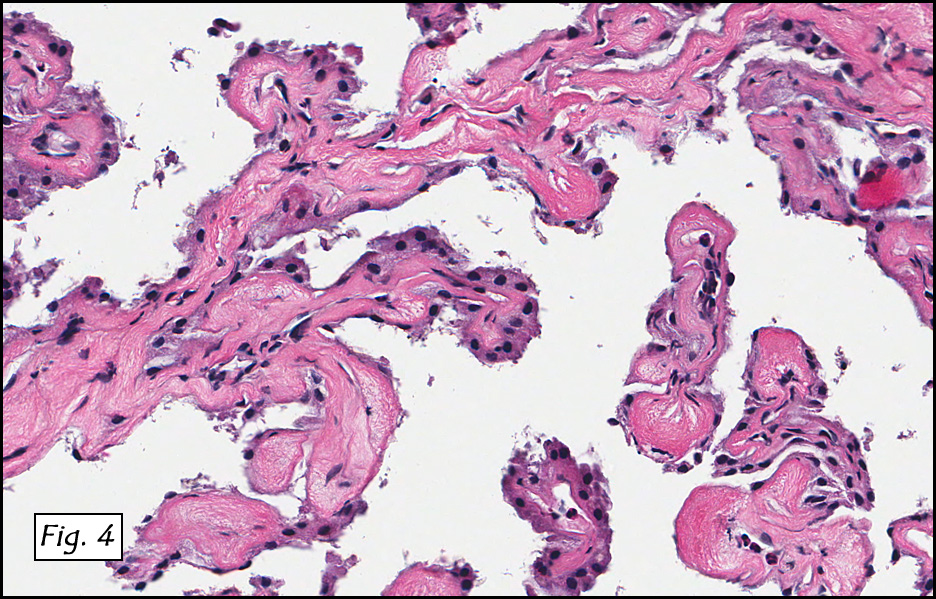
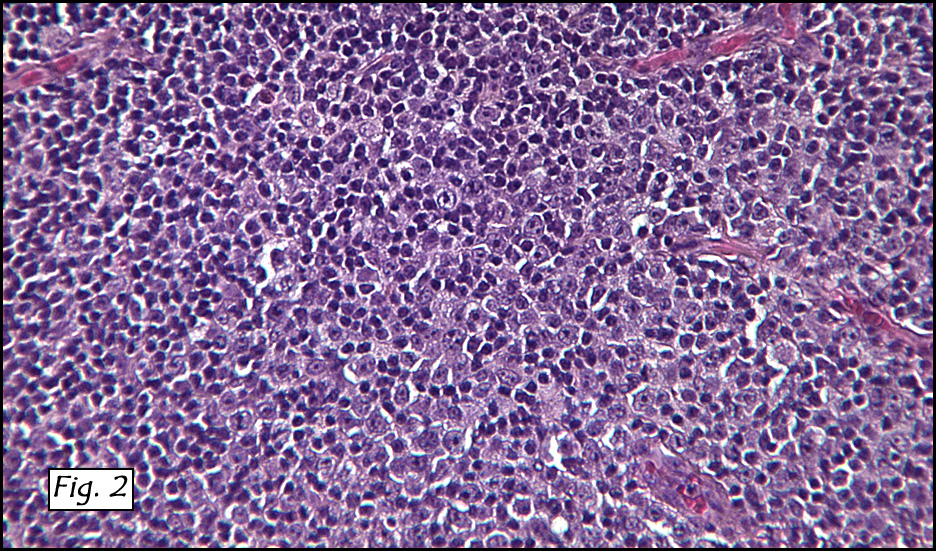
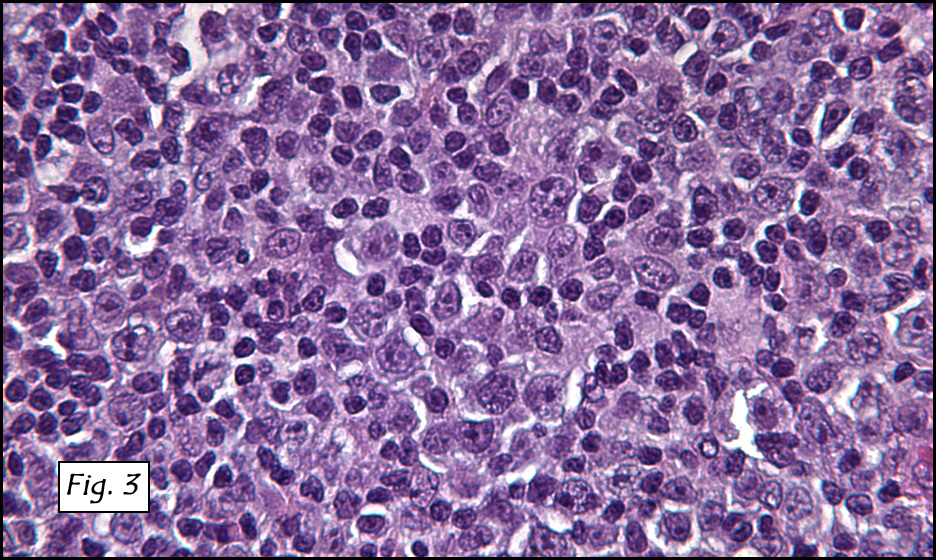
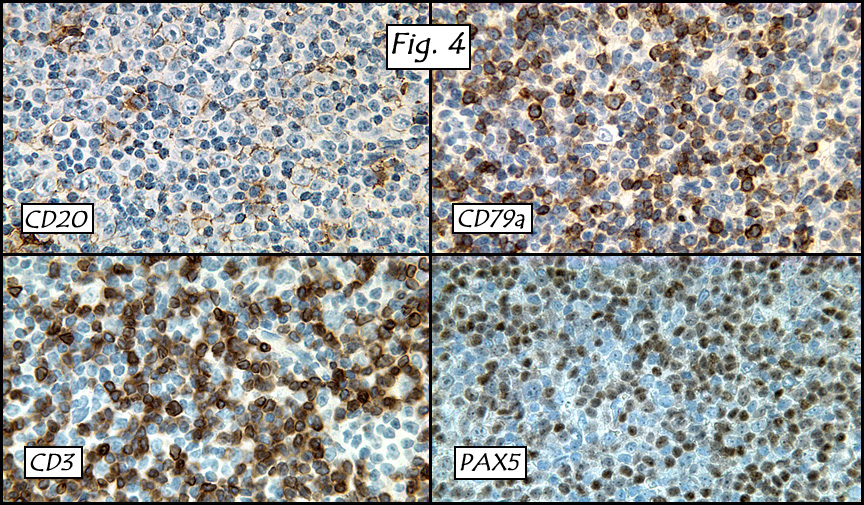
In addition to focal adenomyosis and benign leiomyomata, the uterus was found to have a 3-4 mm superficial tumor in the myometrium consisting of confluent nodules of blunt-ended spindled and epithelioid cells growing in short fascicles around mildly dilated entrapped endometrial glands (Fig. 2). The neoplastic nuclei were round or oval, mildly hyperchromatic and displayed small nucleoli (Fig. 3). Tumor marked for HMB-45 (Fig. 4) as well as for actin, but was negative for CD10. A desmin stain highlighted the surrounding myometrium but was negative in the tumor (Fig. 5). Mitotic figures were only rarely seen (<1/10 HPF). Two pelvic lymph nodes were involved by a proliferation of bland spindled cells surrounding endothelium-lined spaces reminiscent of a pericytoma pattern. Most of the cells had moderate clear to eosinophilic with partially vacuolated cytoplasm (Figs. 6a & 6b). The cells were HMB-45 positive (Fig. 7) and also marked for smooth muscle actin.
The patient had an uneventful postoperative recovery. Additional clinical investigation uncovered stigmata of the tuberous sclerosis complex (TSC). A postoperative Tc99m bone scan showed a solitary focus of increased activity in the upper pole of the left kidney suspicious for angiomyolipoma (AML).
Diagnosis: “Perivascular Epithelioid Cell Tumor (‘PEComa’) of
the Uterus Associated with Lymphangioleiomyoma of the Pelvic Lymph Nodes (another ‘PEComa’) and Cystic Mucinous Tumor of the Ovaryâ€
Mingyi Chen MD, Craig Zuppan MD, and Donald R. Chase MD
Department of Pathology, Loma Linda University and Medical Center, and
California Tumor Tissue Registry, Loma Linda, California
Discussion: The histogenesis of angiomyolipoma, lymphangioleiomyomatosis, and clear cell sugar tumor has long been controversial. The concept of a common cell origin for this collection of neoplasms was first advanced by Bonetti et al. in a letter published in the American Journal of Surgical Pathology in 1992, in which the authors noted that both angiomyolipoma (AML) and clear cell ‘‘sugar’’ tumor of the lung (CCST) were immunoreactive with melanocytic markers and exhibited an epithelioid appearance with clear-acidophilic cytoplasm, usually in a perivascular distribution. In 1996, Pea et al proposed the perivascular epithelioid cell (PEC) as the cell of origin of these tumors and noted that due to the pleuri-immunohistochemical potential of the PEC, that tumors of this type may express varying degrees of melanocytic, muscle, pericytic, and perivascular epithelioid differentiation. Since then, the World Health Organization has broadly defined PEComas as ‘‘mesenchymal tumors composed of histologically and immunohistochemically distinctive perivascular epithelioid cells.’’
The PEComa family of tumors is now recognized to include angiomyolipoma (AML), lymphangioleiomyomatosis (LAM), clear-cell “sugar†tumor of the lung (CCST), and clear-cell myomelanocytic tumor of the falciform ligament/ligamentum teres. The PEC is also found in other clear-cell tumors such as “abdominopelvic sarcoma of perivascular epithelioid cellsâ€, “primary extrapulmonary sugar tumor,†and “extra-renal epithelioid AMLâ€.
PEComas have been reported in almost every body site, and the growing list includes gynecological sites, genitourinary and gastrointestinal locations, extremities, skin, as well as isolated reports in the heart, breast, oral cavity, orbit, and skull base. The uterus is the most commonly reported site of a PEComa.
Although the clinical significance is unclear, Vang and Kempson have divided uterine PEComas into two types based on morphology and immunohistochemical characteristics:
• Type A tumors show a tongue-like growth pattern and are composed of cells with clear to eosinophilic pale granular cytoplasm, diffuse HMB-45 expression, and focal smooth muscle reactivity.
• Type B tumors are composed of epithelioid cells with less prominent clear cell features, only a few of which are HMB-45 positive. In addition, these cells demonstrate extensive muscle marker expression and a lesser degree of tongue-like growth pattern.
The PEC does not have a known normal histologic counterpart. Morphologically it is somewhat spindled, yet epithelioid with clear to granular cytoplasm, a round to oval centrally located nucleus and an inconspicuous nucleolus. Immunohistochemically, PEC proliferations co-express both myogenic markers (actin and desmin) and melanocytic markers such as HMB45, HMSA-1, MelanA/Mart1, and microophthalmia transcription factor (MiTF). It minimally decorates for vimentin. Ultrastructure shows PEC tumors to contain microfilament bundles with electron-dense condensations, numerous mitochondria and membrane-bound dense granules.
Many PEComas have been reportedly associated with the tuberous sclerosis complex (TSC). TSC is an inherited disorder characterized by mutations of TSC1 and TSC2, two tumor suppressor genes located on chromosomes 9q34 and 16p13.3, respectively. This chromosomal imbalance has been demonstrated both in TSC and in PEComa. The TSC1 gene produces hamartin, whereas the TSC2 gene produces tuberin, these two proteins being responsible for the regulation of multiple intracellular signaling pathways of cell growth control. Inherited or acquired loss of heterozygosity at the TSC1 and TSC2 loci leads to uncontrolled cell proliferation and the development of hamartomas and tumors. Activation of the mTOR signal pathway through loss of inhibition by TSC1/2 likely occurs in most, if not all PEComas.
PEComas Versus Uterine Smooth Muscle Cell Tumors (U-SMTs). PEComas may be confused with true smooth muscle neoplasms with both spindled and epithelioid morphology. On microscopic examination, PEComas show overlapping features with epithelioid U-SMTs, as they have cells with abundant clear or eosinophilic cytoplasm and round to oval nuclei arranged in sheets, small solid nests, or cords separated by scant hyalinized stroma. Features that will favor the diagnosis of PEComa over an epithelioid U-SMT include the association with tuberous sclerosis and lymphangiomyomatosis, the presence of multinucleated giant cells and ‘‘spiderlike’’ cells in PEComas, and the expression of melanocytic markers such as HMB-45, Melan A, and MiTF. Other helpful clues include the absence of delicate capillary networks in epithelioid SMT and the frequent keratin and EMA expression in epithelioid U-SMTs but none in PEComas.
PEComas Versus Uterine Tumor Resembling an Ovarian Sex Cord Tumors (UTROSCT). UTROSCTs are rare stromal tumors showing prominent sex cord–like differentiation. On microscopic examination, the neoplastic cells show an epithelioid appearance with indistinct eosinophilic (sometimes vacuolated) cytoplasm and oval to round nuclei and grow in tight nests, cords, and sheets. Sometimes, spindle-shaped cells are present, and the stroma is generally sparse with some hyaline strands. The epithelial differentiation in UTROSCTs is more pronounced than in PEComas, with tubular formation, retiform differentiation, or prominent vacuolated cytoplasm, as seen in the luteinized cells of sex cord stromal tumors of the ovary. The presence of sex cord stromal markers (inhibin and calretinin) in UTROSCTs is helpful in this differential diagnosis.
PEComas Versus Placental Site Trophoblastic Tumor and Epithelioid
Trophoblastic Tumor. Microscopically, placental site trophoblastic tumors (PSTTs) and epithelioid trophoblastic tumors (ETTs) are composed of mononucleated round or polyhedral intermediate trophoblastic cells with abundant eosinophilic to clear cytoplasm frequently associated with a diffuse or nested growth. Features favoring a diagnosis of trophoblastic tumor include history of recent pregnancy or abortion and an elevated serum human chorionic gonadotropin level, an infiltrative growth of single cells or small aggregates of cells dissecting individual muscle fibers (PSTT), prominent vascular involvement with associated fibrinoid change (PSTT), islands or nests of cells surrounded by extensive necrosis or a hyaline-like matrix (ETT), as well as immunoreactivity for inhibin, human placental lactogen, and p63 (ETT), with negativity for myomelanocytic markers.
PEComas Versus Endometrial Stromal Sarcoma. Cells with abundant dense eosinophilic cytoplasm have been rarely described in endometrial stromal tumors, and this finding may lead to a differential diagnosis of a PEComa, especially in small samples. In these cases, the distinct immunohistochemical features (CD10 vs HMB-45) are helpful clues for the diagnosis.
PEComas Versus Melanoma. Although unusual, conventional clear cell sarcoma (melanoma of soft tissue) or metastatic melanoma from other genital or extragenital sites could involve the uterus and should always be in the differential diagnosis of PEComas. Diffuse positivity for S100, HMB-45, and Mart-1, and negative expression of smooth muscle markers will favor the diagnosis of melanoma. Identification of the t(12; 22) (EWS-ATF1) translocation is diagnostic for CCS.
Our presentation case shows a most interesting combination of two PEComas in the setting of the tuberous sclerosis complex and points to the possibility of “PEComatosisâ€, a condition that is likely under-reported and therefore under-studied. Literature supports the existence of malignant PEComas that have metastasized, but to date, no uterine primary tumor has been described with this behavior, and virtually all metastasizing lesions have been malignant angiomyolipomas (Pea M, et. al, and Vip SK, et. al). For this reason, we interpret the multicentricity of these two PEComas as being synchronous primaries.
It has been suggested that HMB-45 immunostaining should be performed on uterine mesenchymal tumors with lymphangiomyomatous pattern or with clear and epithelioid features to identify PEC tumors, and that the diagnosis of a PEComa warrants the examination of the patient for TSC.
Although most uterine PEComas follow a benign course, they should be viewed as having uncertain malignant potential particularly those larger than 8 cm with marked hypercellularity, cytological atypia, high mitotic activity (including atypical forms), coagulative necrosis and/or an infiltrative growth pattern. The mainstay of treatment of conventional uterine PEComas is wide excision or hysterectomy. A recent pilot study from the Dana-Farber Cancer Institute suggests that malignant PEComas may show responses to mTOR Inhibitor (Sirolimus) treatment.
Suggested Reading:
Martignoni G, Pea M, Reghellin D, Zamboni G, Bonetti F. PEComas: the past, the present and the future. Virchows Arch. 2008 Feb;452(2):119-32.
Folpe AL, Mentzel T, Lehr HA, Fisher C, Balzer BL, Weiss SW. Perivascular epithelioid cell neoplasms (PEComas) of soft tissue and gynecologic origin: a clinicopathologic study of 24 cases. Mod Pathol 2005;18:48A.
Folpe AL. Neoplasms with perivascular epithelioid cell differentiation (PEComas) In: Fletcher CDM, Unni KK, Epstein J, Mertens F (eds). Pathology and Genetics of Tumours of Soft Tissue and Bone. Series: WHO Classification of tumours. IARC Press: Lyon, 2002, pp. 221-222.
Vang R, Kempson RL. Perivascular epithelioid cell tumor (‘PEComa’) of the uterus: a subset of HMB-45-positive epithelioid mesenchymal neoplasms with uncertain relationship to pure smooth muscle tumors. Am J Surg Pathol 2002;26:1-13.
Pea M, Bonetti F, et. al. Apparent renal cell carcinomas in tuberous sclerosis are heterogeneous; the identification of malignant epithelioid angiomyolipoma. Am J Surg Pathol 1998:22:180-187.
Vip SK, Sim CS, Tan BS. Liver metastasis and local recurrence after radical nephrectomy for an atypical angiomyolipoma. 2001:J Urol 165:898-899.
 History: A previously healthy 80 year-old man presented with a one-month history of right axillary lymphadenopathy. Physical examination revealed a mildly tender, firm, two cm lymph node in the right axilla. No other lesions identified.
History: A previously healthy 80 year-old man presented with a one-month history of right axillary lymphadenopathy. Physical examination revealed a mildly tender, firm, two cm lymph node in the right axilla. No other lesions identified.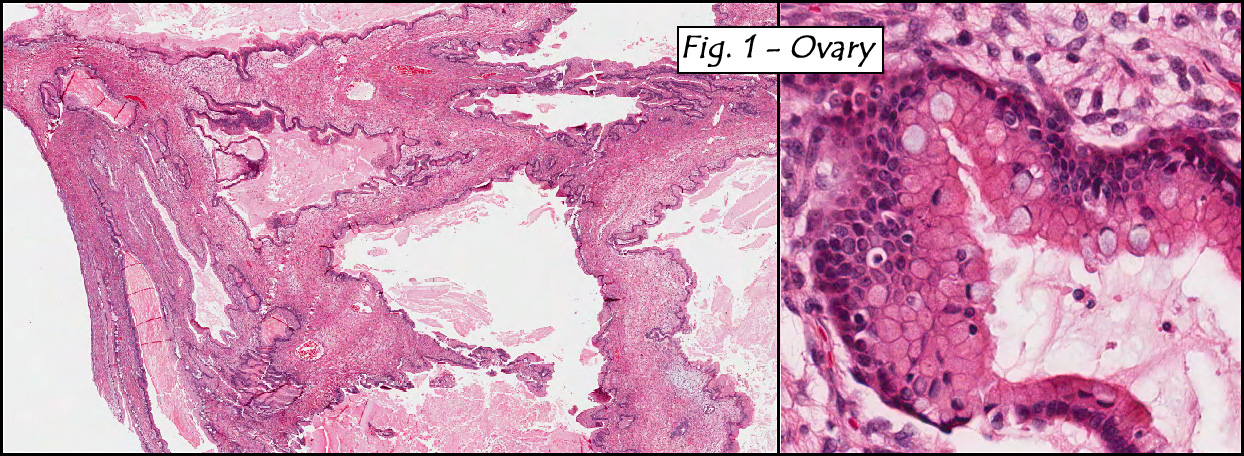{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}