History: A 59-year-old man had a cutaneous melanoma of his right abdomen excised three years earlier. He presented with an ulcerated, enlarging scar at the site of previous excision.
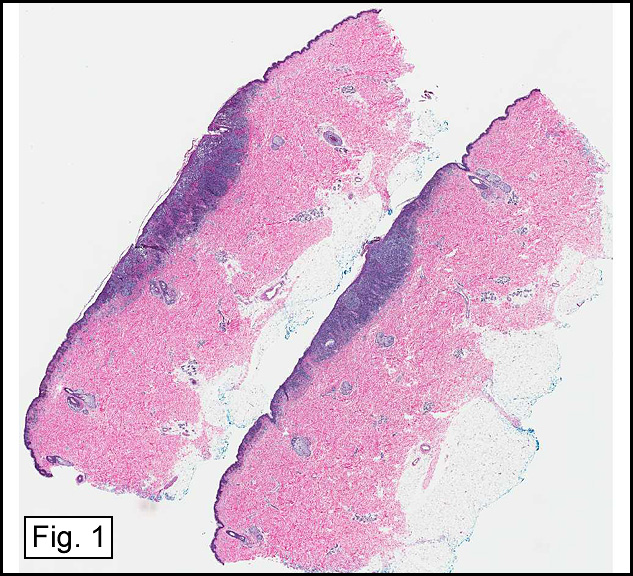
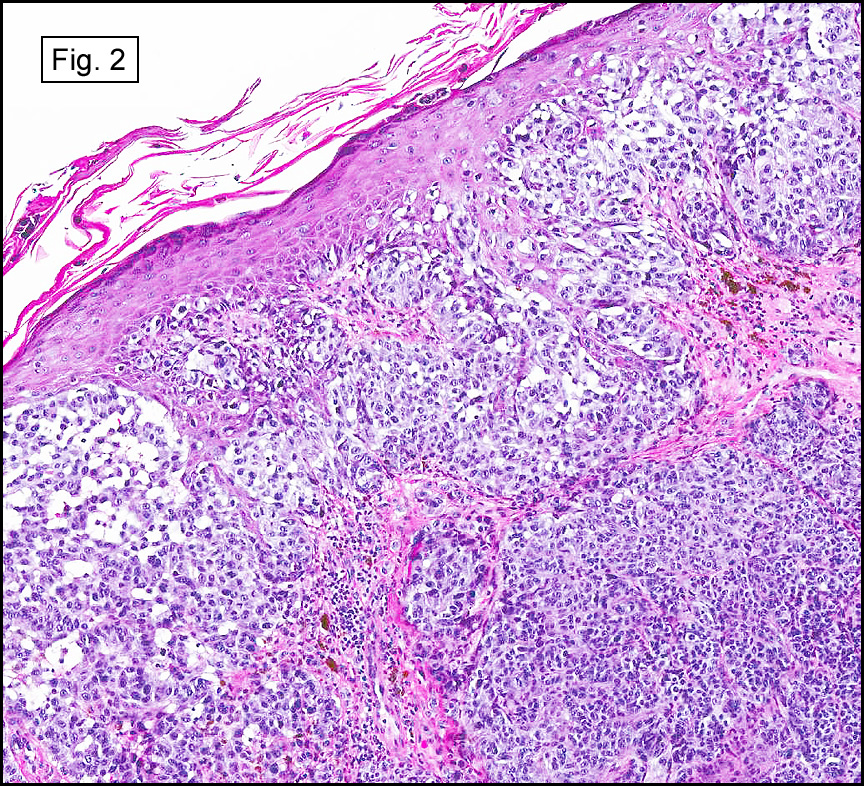
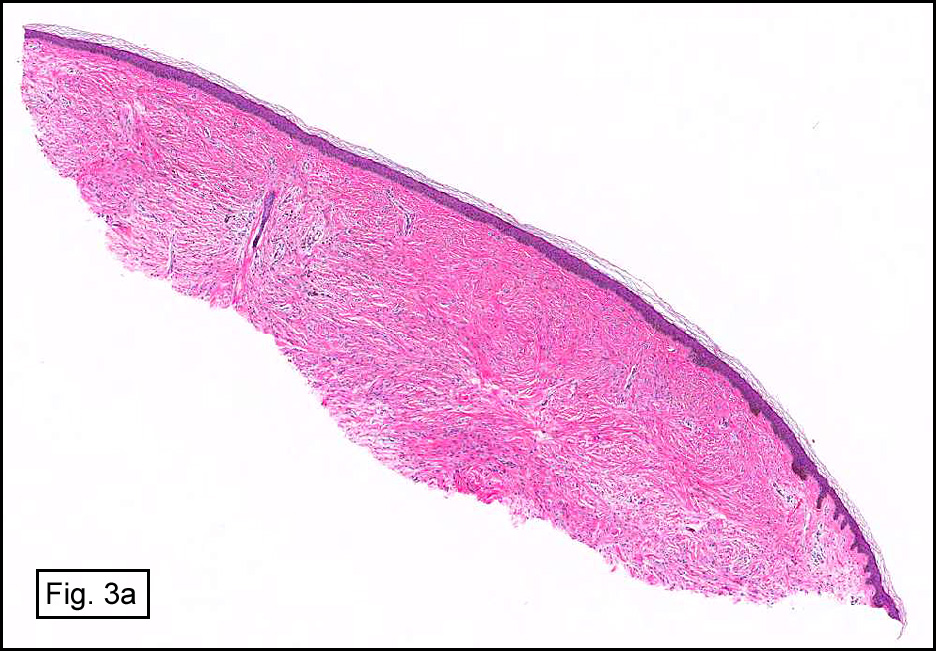
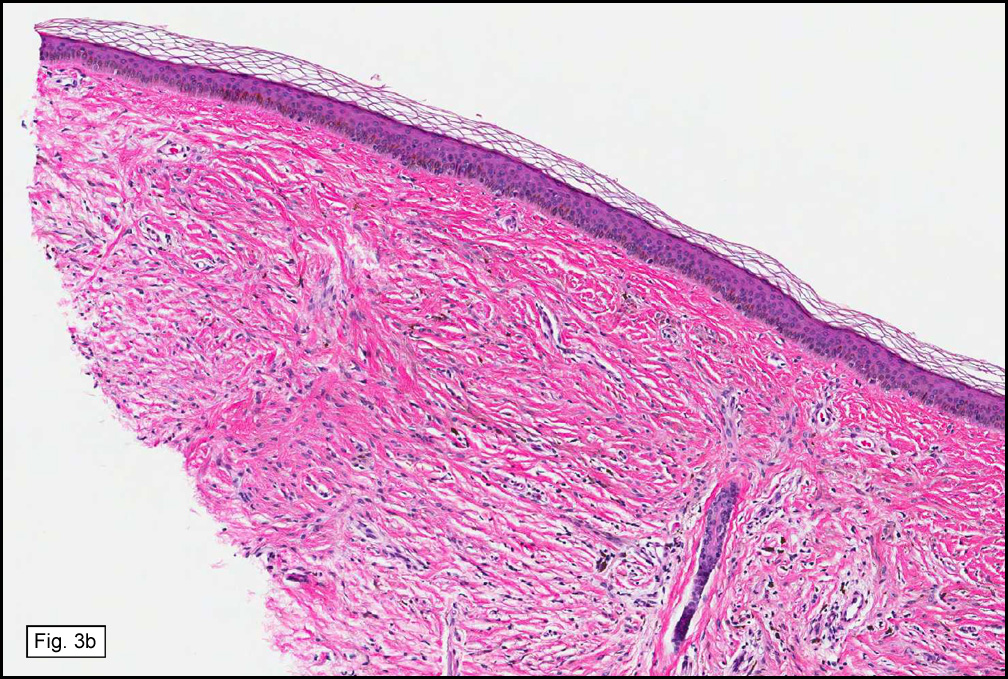
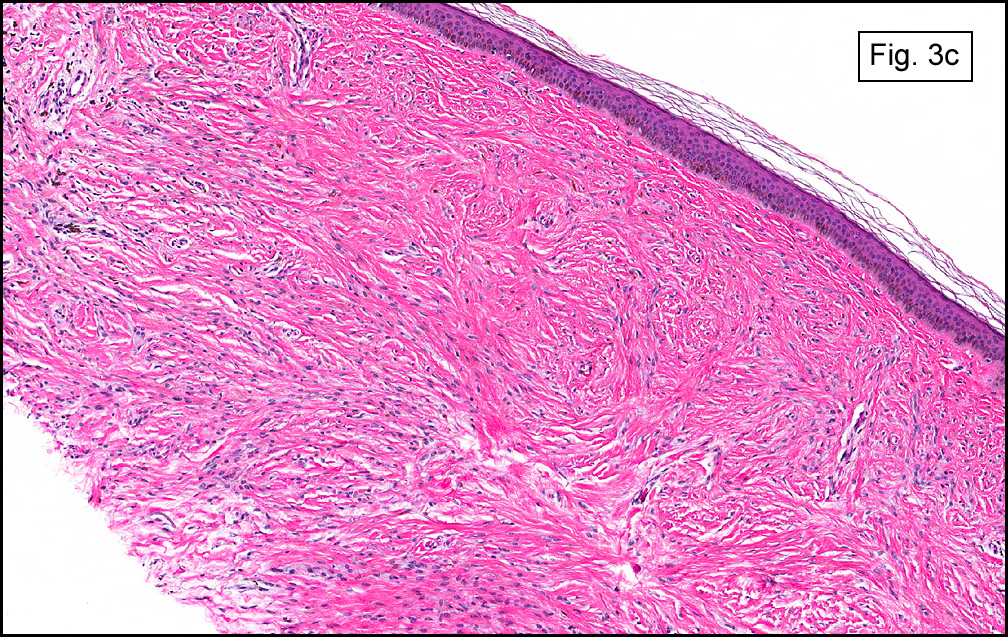
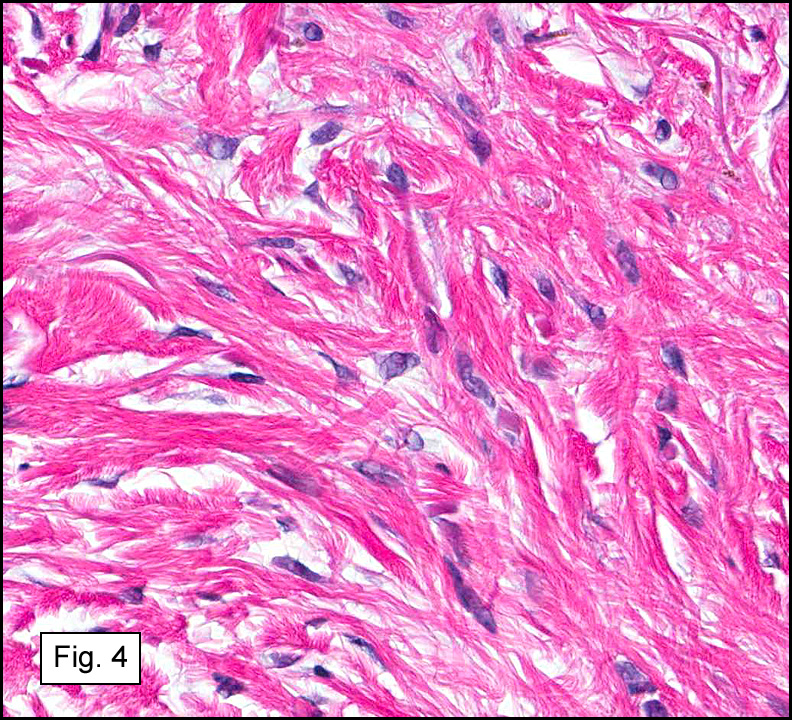
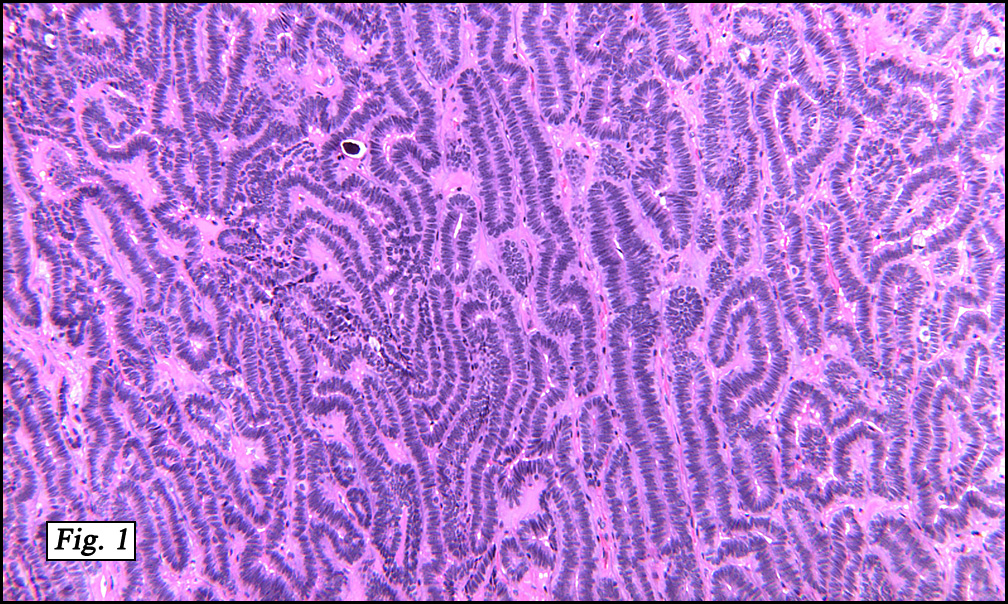
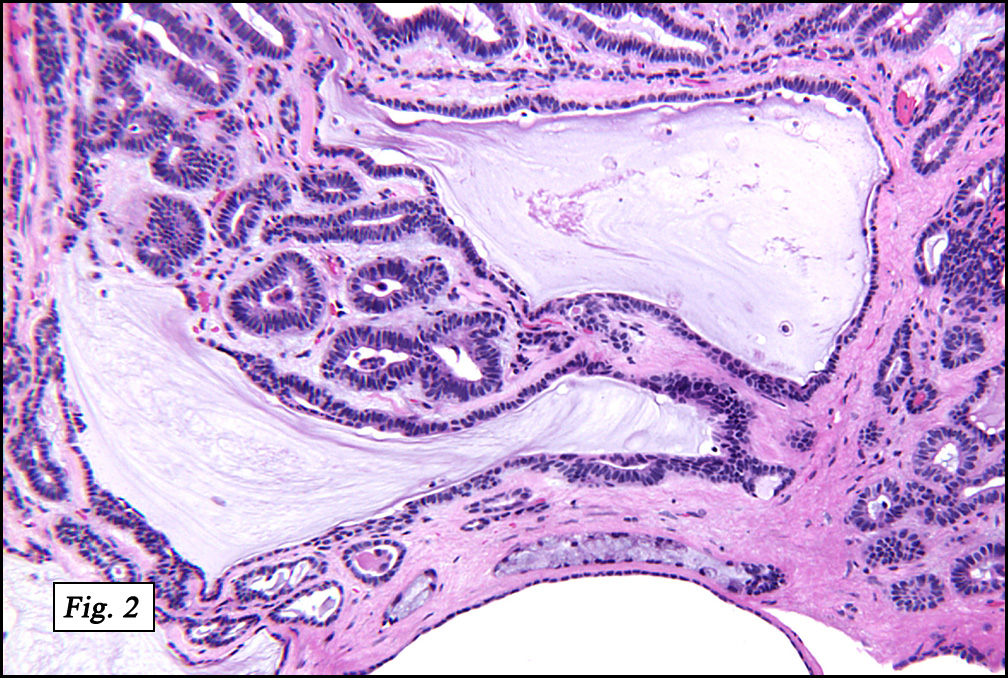
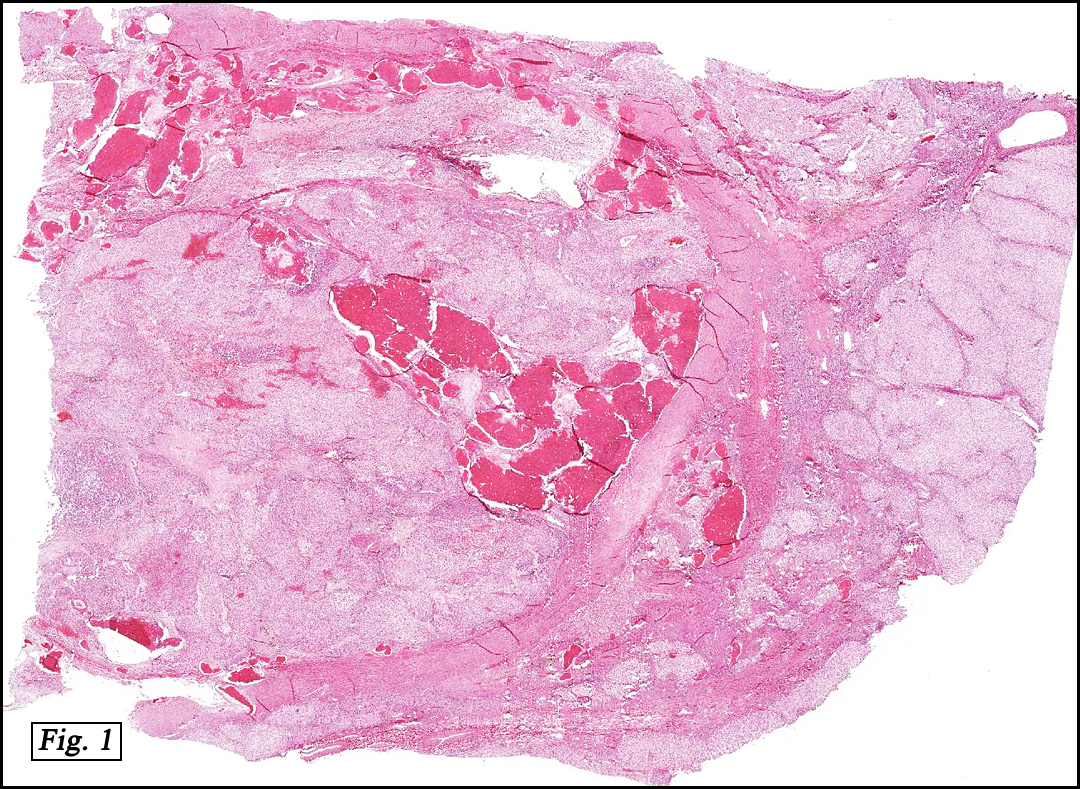
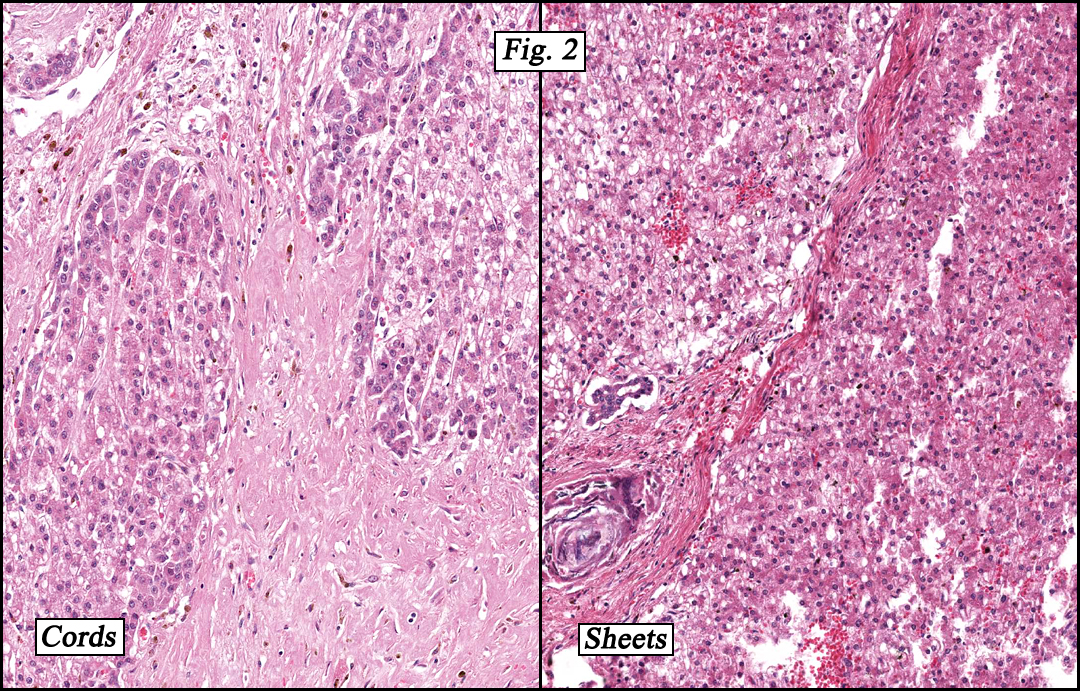
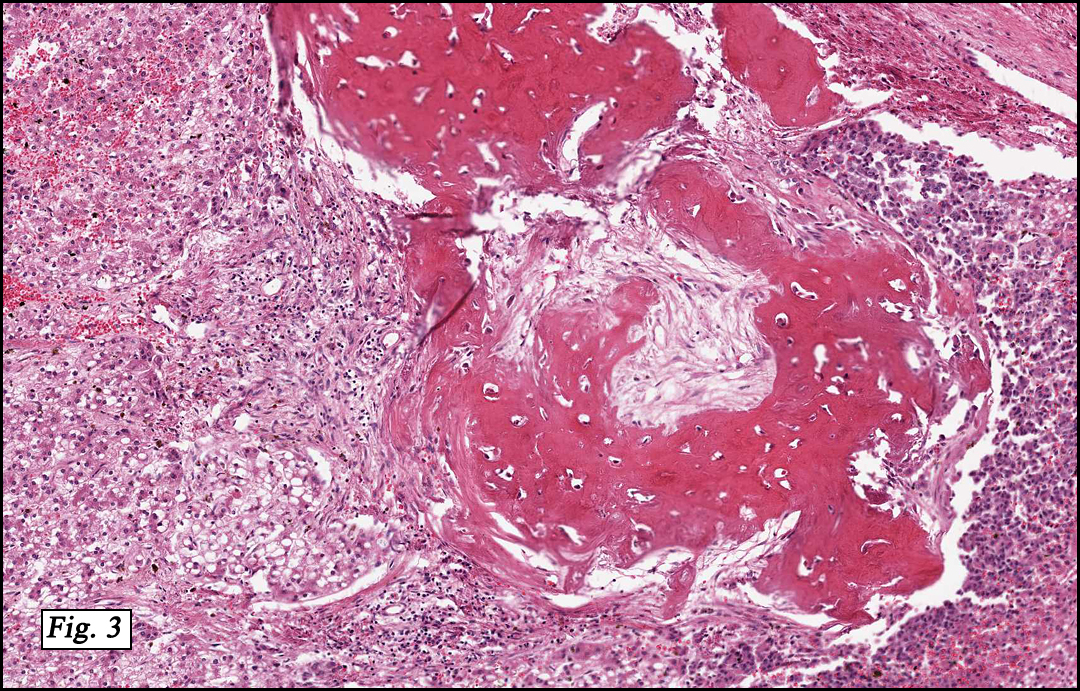
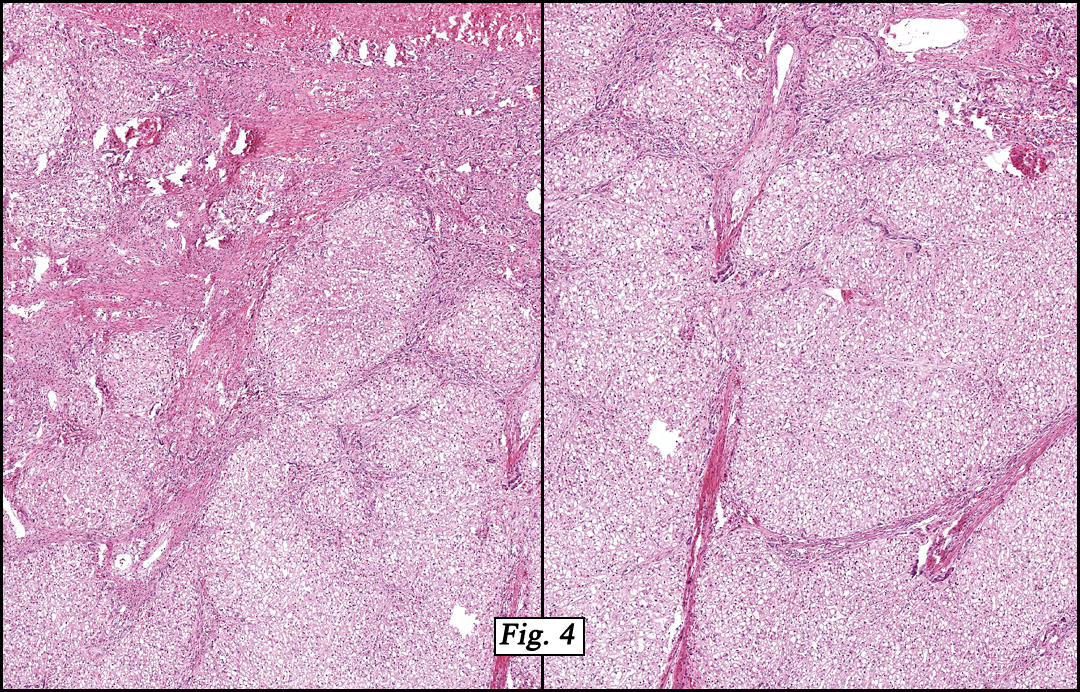
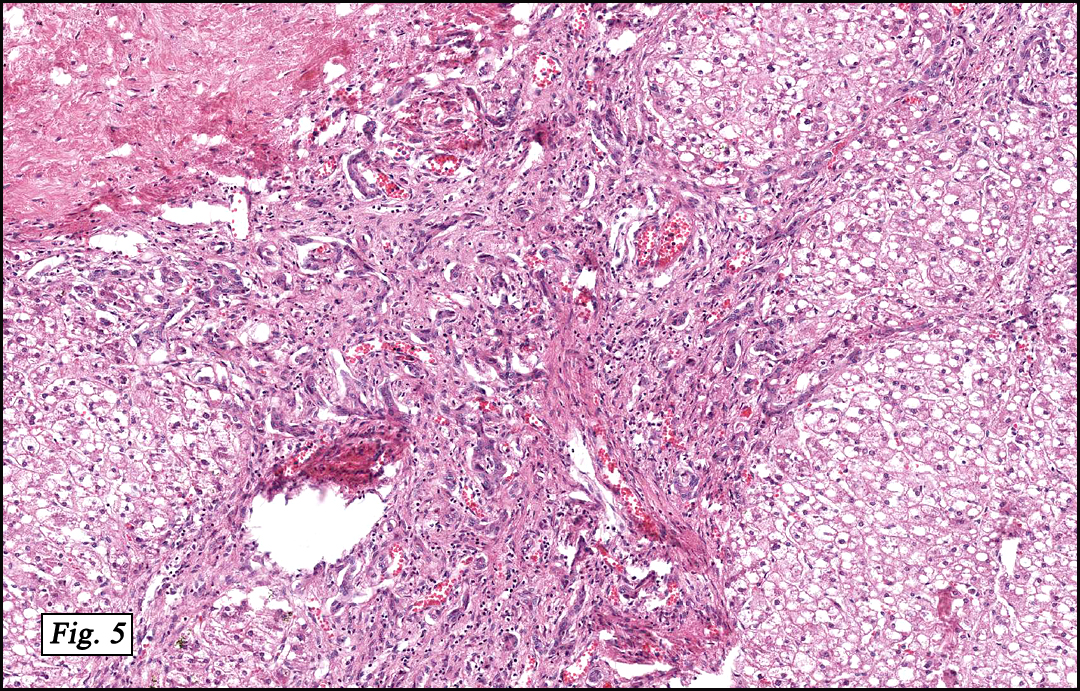
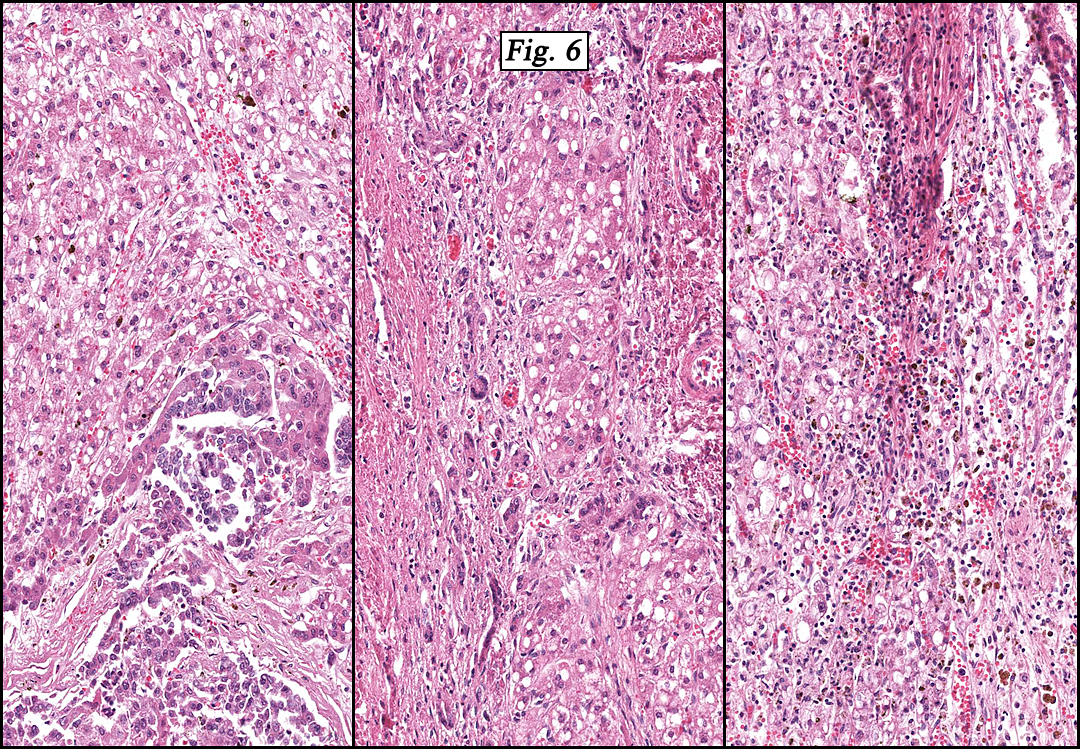
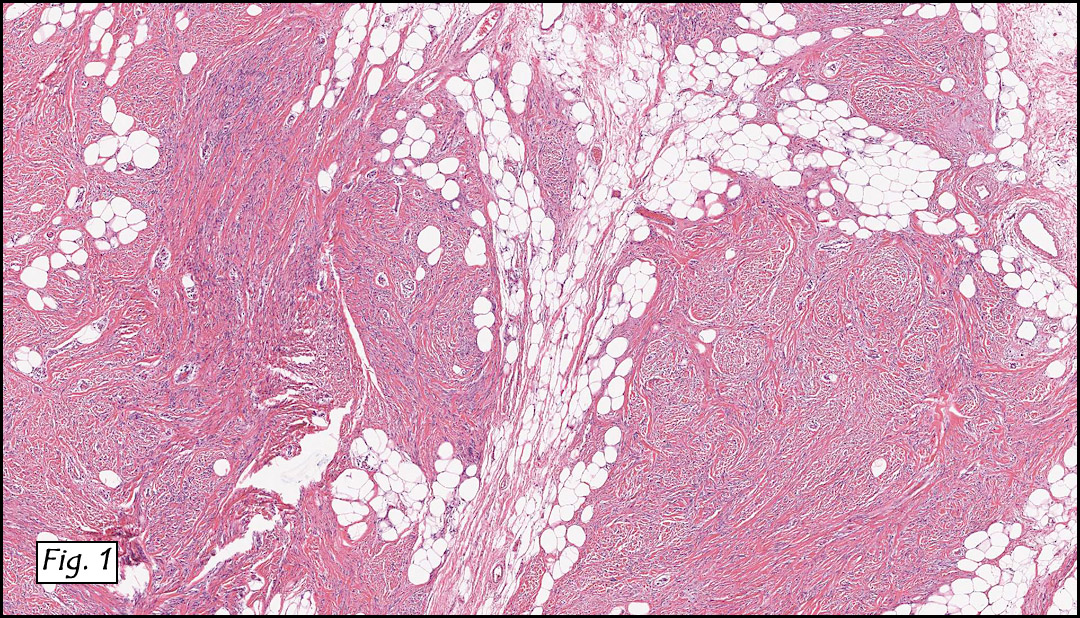
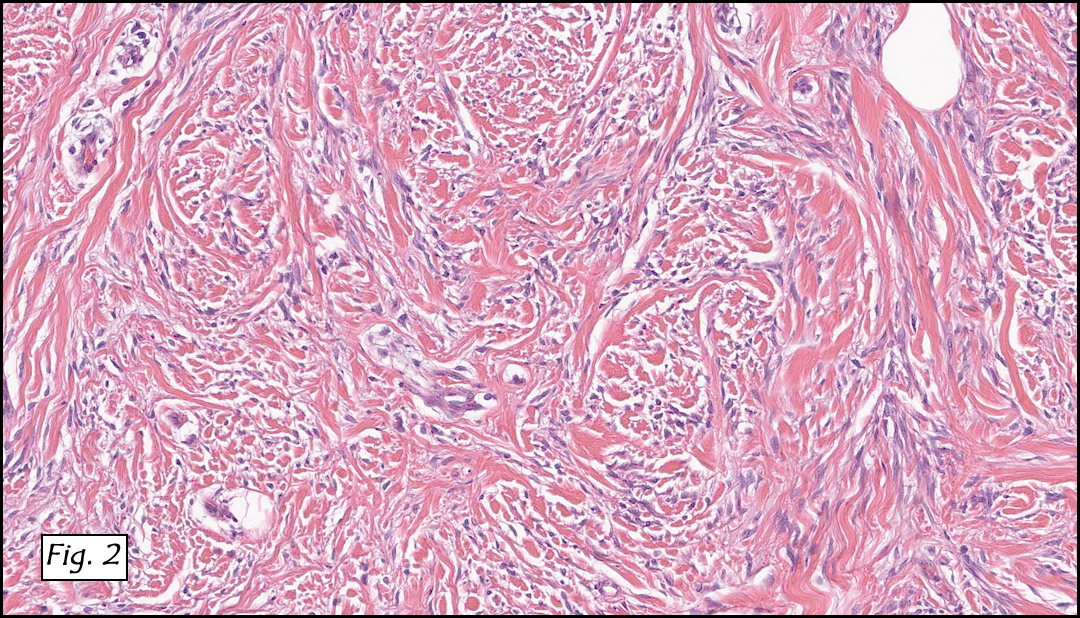
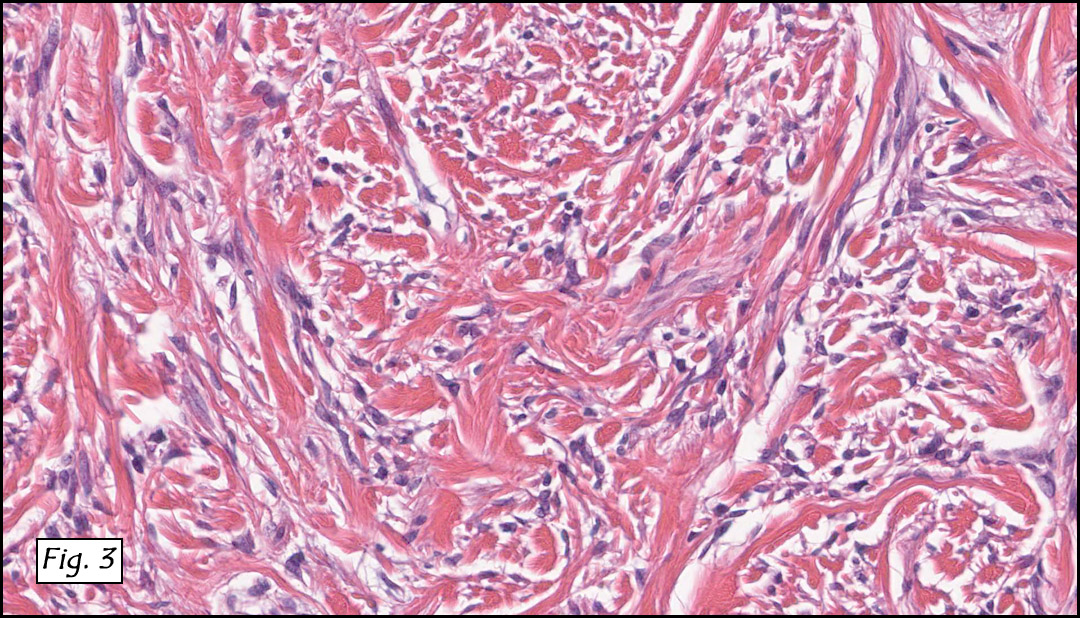
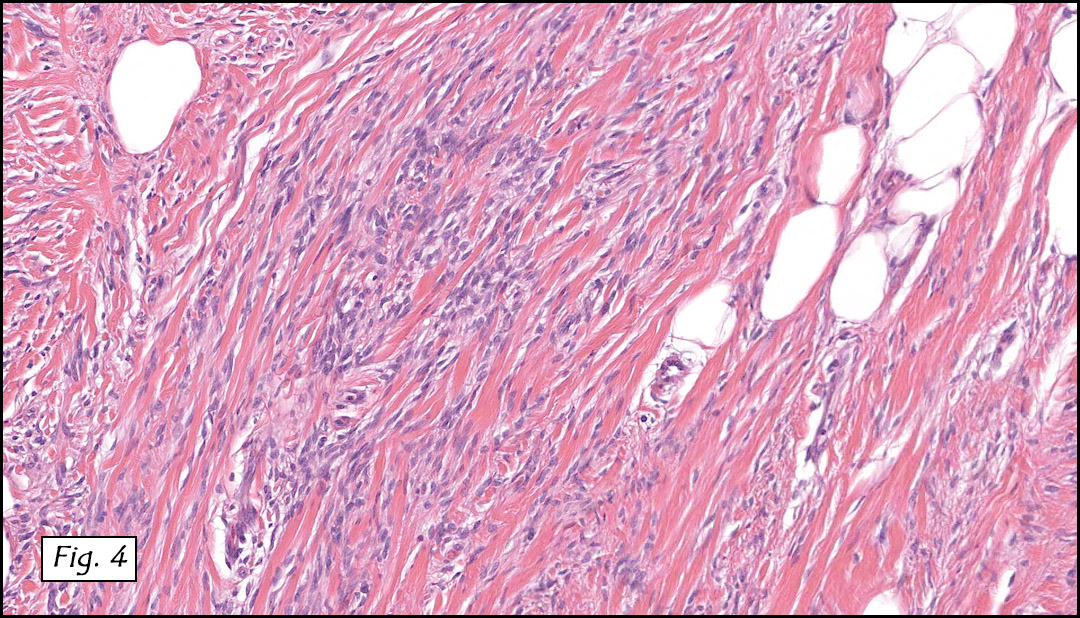
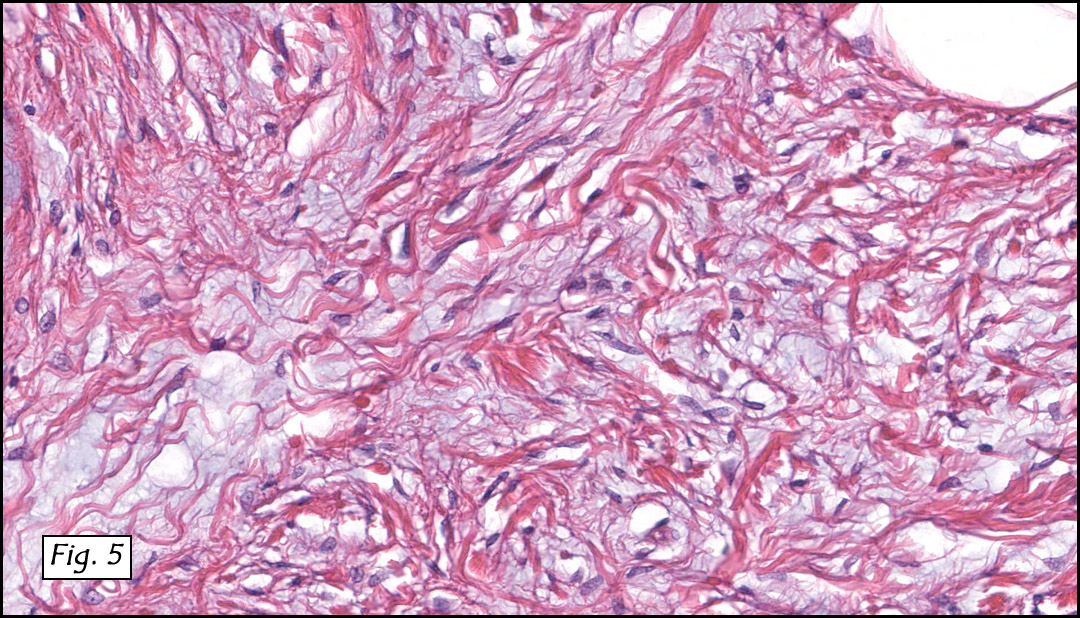
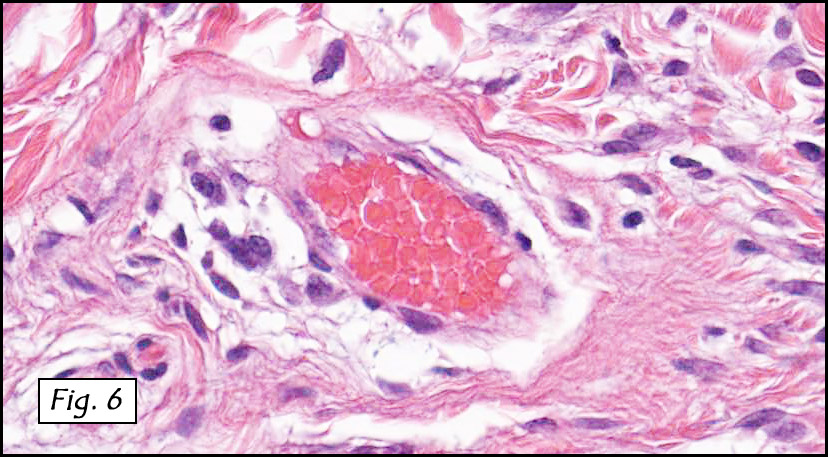
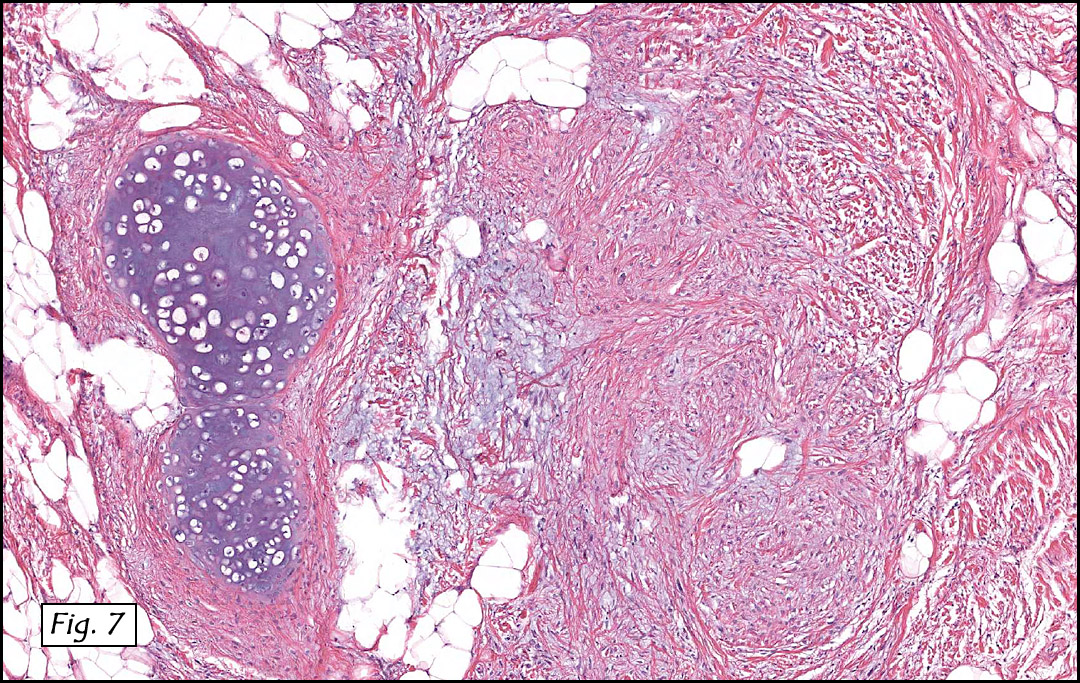
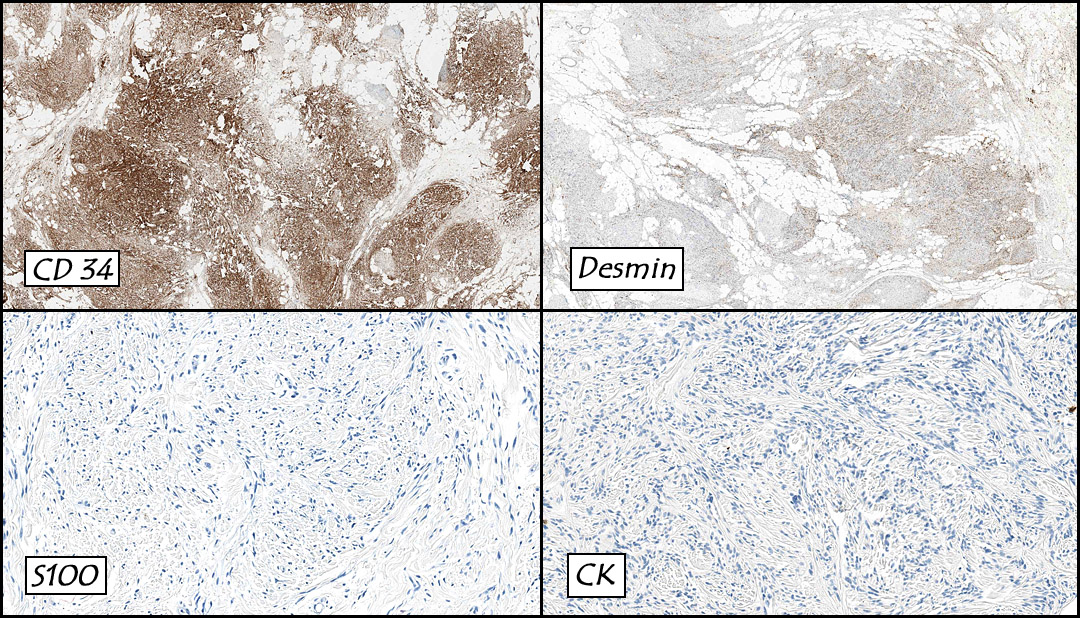
The original melanoma was classic, composed mostly of polygonal melanocytes, without spindled elements (Figs. 1, 2). The peripheral margins were microscopically free of tumor (Fig. 1). The more recent excision had a cicatrix appearance (Figs. 3a,3b,3c). Upon closer examination, the tumor had an infiltrative pattern consisting most of fibrosing spindle cells (Fig. 4). The cytology of these spindle cells was rather bland, resembling fibroblasts. Interspersed between the spindle cells were (rarely) macrophages filled with dark pigment (Fig. 5). Polygonal cells, which dominated in the original tumor, were not present. Immunohistochemical stains showed the spindled cells to be positive for both HMB-45 (Fig. 6 left) and Melan A (Fig. 6 right).
Diagnosis: “Recurrent Melanoma with Desmoplastic Phenotype (“Desmoplastic Melanomaâ€)
Jonathan Zumwalt, MSIV, Resa L. Chase, MD, and Donald R. Chase, MD
Department of Pathology and Human Anatomy, Loma Linda University and Medical
Center, Loma Linda, California
California Tumor Tissue Registry, Loma Linda, California
Discussion: In 1812, Rene Laennec first described the disease entity currently known as “melanomaâ€, in a paper entitled “The Melanosesâ€. Today melanoma is seen in 1/39 Caucasian men and 1/58 Caucasian women in the United States. There are approximately 70,000 cases of invasive melanoma diagnosed every year and almost 9,000 lives are lost every year to this tumor. The incidence of melanoma has markedly increased over the past 10-20 years and may be attributed to three non-biologic factors: 1) increased rates of screening, 2) an increase in the number of biopsies being taken and 3) changes involving histologic interpretation of early evolving lesions. The main risk factors for developing melanoma are: family history, history of atypical nevi, high nevus count, history of increased sun exposure and physical traits (i.e. light colored hair and eyes).
There are a variety of factors to consider when reporting a melanoma:
• Breslow thickness is the most important factor for melanoma staging.
• Ulceration is now the second most powerful predictor of survival.
• Mitotic activity of the tumor shows a correlation between replication and survival.
• Radial and vertical growth phases may help characterize the tumor’s aggressiveness. Tumor infiltrating lymphocytes seem to indicate the presence of vertical growth phase.
• Microscopic satellites cut down the five year survival rate of a patient by more than half.
• Regression is still controversial and experts give different opinions on this phenomenon.
• The anatomic primary tumor site of a melanoma does correlate with patient survival.
• Clark’s levels are no longer used in today’s practice for staging, but many physicians still report them.
Melanomas today are divided into four common variants: superficial spreading, lentigo maligna, acral lentiginous, and nodular.
1. Superficial spreading melanomas make up 75% of melanomas. Histologically, they are asymmetric, poorly circumscribed and lack cellular maturation. Upward invasion of melanocytes into the epidermis is characteristic, as is a prolonged radial growth phase.
2. Lentigo maligna melanomas begin as tan-brown macule on sun damaged skin that evolves into larger, darker areas with asymmetric foci. Histologically, the neoplastic melanocytes are arrayed along the dermal-epidermal junction in a lentiginous pattern against a background of epidermal atrophy and solar elastosis.
3. Acral lentiginous melanomas commonly arise on the palmar, plantar, subungual, and occasionally, mucosal surfaces and typically occur in dark skinned individuals. Histologically, they are characterized by a lentiginous array of atypical melanocytes along the dermal-epidermal junction, with foci of confluent melanocytic growth and progress to large junctional nests that are composed of atypical melanocytes.
4. Nodular melanomas are darkly pigmented polypoid or pedunculated nodules. Histologically, these typically show dermal growth in isolation, but occasionally, in association with an epidermal component.
One rare variant of melanoma is desmoplastic melanoma (DM). It is frequently mistaken for a scar, dermatofibroma and/or sarcoma. Grossly DM generally appears as an amelanotic, pale, and fleshy nodule or papule, usually resembling a scar. A variety of studies have shown that it is highly associated with lentigo maligna (24 percent). Histologically these tumors show a poorly circumscribed vertical growth phase tumor with spindle-shaped malignant cells with a fibrogenic stromal pattern. The nuclei of these cells are typically elongated and hyperchromatic. The morphology of these cells resemble fibroblasts. Immunohistochemical staining usually shows S-100 positivity, while HMB-45, Melan-A, tyrosinase and microphthalmia transcription factor are generally negative.
There has been controversy regarding the prognosis of DM. Original studies found it a “highly malignant, stubbornly recurring and often metastasizing neoplasm.†In the past twenty years this perception has started to change, allowing for an indolent behavior. The confusion of this may be due to the failure of some studies to detect differences between DM and conventional melanoma. A second reason for the conflicting results is the heterogeneity among melanomas declared as desmoplastic.
Differential Diagnosis:
Sclerosing Melanocytic Nevus usually occurs in younger patients, and usually in skin which has not been actinically damaged. Histologically seen from low power, they are sometimes perceived as a circumscribed “silhouette†with little or no cytologic atypia or mitotic activity. In contrast, DM, tends to be asymmetric, infiltrative and poorly circumscribed. Most DMs are associated with a previous or current in-situ melanoma.
Dermal Scar or Dermatofibroma: Without evidence of abnormal melanocytic changes in the epidermis, recognition of a dermal spindle cell proliferation can be most difficult to identify as melanoma. Often DM may be mistaken for a scar. But fibroblasts in scars typically are arranged in parallel to the skin surface, while blood vessels are perpendicular. Dermatofibromas tend to have spindle cells that wrap around the collagen bundles. In contrast, DM has fusiform cells oriented both parallel and perpendicular to the skin surface. Also, DM usually shows some cytologic atypia with elongated hyperchromatic nuclei.
Sarcomas or Sarcomatoid Carcinoma: DM at times can resemble a spindle cell sarcoma, and immunohistochemistry is usually needed to help distinguish these differences. The use of S-100, muscle markers and epithelial markers can help distinguish DM from desmoplastic carcinoma, leiomyosarcoma, pleomorphic sarcoma and malignant peripheral nerve sheath tumor. Sarcomatoid carcinoma usually stains strongly for 34BE12, which does not stain melanomas.
Suggested Reading:
Busam KJ. Desmoplastic Melanoma. Clin Lab Med. 2011 Jun;31(2):321-30.
Divito SJ, Ferris LK. Advances and short comings in the early diagnosis of melanoma. Melanoma Res. 2010 Dec;20(6):450-8.
Garbe C, Eigentler TK, Bauer J, et al. Histopathological diagnostics of malignant melanoma in accordance with the recent AJCC classification 2009: Review of the literature and recommendations for general practice. J Dtsch Dermatol Ges. 2011 Jun 9. doi: 10.1111/j.1610-0387.2011.07714.x.
Kong Y, Kumar SM, Xu X. Molecular pathogenesis of sporadic melanoma and melanoma-initiating cells. Arch Pathol Lab Med. 2010 Dec;134(12):1740-9.
Leong SP, Gershenwald JE, Soong SJ, Schadendorf D, Tarhini AA, Agarwala S, Hauschild A, Soon CW, Daud A, Kashani-Sabet M. Cutaneous melanoma: a model to study cancer metastasis. J Surg Oncol. 2011 May 1;103(6):538-49. doi: 10.1002/jso.21816.
Murali R, Zannino D, Synnott M, McCarthy SW, Thompson JF, Scolyer RA. Clinical and pathological features of metastases of primary cutaneous desmoplastic melanoma. Histopathol. 2011 May;58(6):886-95. doi: 10.1111/j.1365-2559.2011.03808.x. Epub 2011 Mar 25.
Murali R, Shaw HM, Lai K, McCarthy SW, Quinn MJ, Stretch JR, Thompson JF, Scolyer RA. Prognostic factors in cutaneous desmoplastic melanoma: a study of 252 patients. Cancer. 2010 Sep 1;116(17):4130-8.
Piris A, Mihm MC Jr, Duncan LM. AJCC melanoma staging update: impact on dermatopathology practice and patient management. J Cutan Pathol. 2011 May;38(5):394-400. doi: 10.1111/j.1600-0560.2011.01699.x. Epub 2011 Mar 9.
Sade S, Al Habeeb A, Ghazarian D. Spindle cell melanocytic lesions–part I: an approach to compound naevoidal pattern lesions with spindle cell morphology and Spitzoid pattern lesions. J Clin Pathol. 2010 Apr;63(4):296-321.
Sade S, Al Habeeb A, Ghazarian D. Spindle cell melanocytic lesions: part II–an approach to intradermal proliferations and horizontally oriented lesions. J Clin Pathol. 2010 May;63(5):391-409.
Whiteman DC, Pavan WJ, Bastian BC. The Melanomas: A synthesis of epidemiological, clinical, histopathological, genetic, and biological aspects, supporting distinct subtypes, causal pathways, and cells of origin. Pigment Cell Melanoma Res. 2011 Jun 27. doi: 10.1111/j.1755-148X.2011.00880.x.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}