History: A 60-year-old woman presented with a urinary tract infection. Past medical history was positive for recently-diagnosed lymphoma status post chemotherapy. Cystoscopy showed a small polypoid mass in the trigone of the bladder.
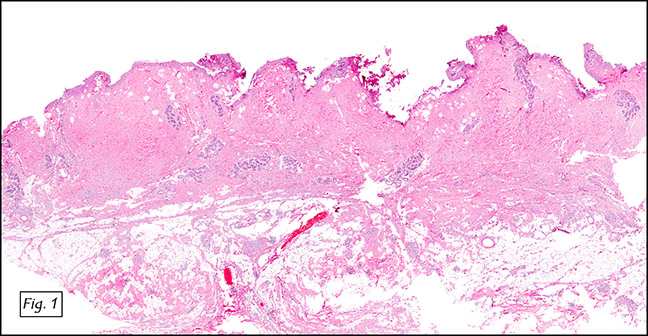
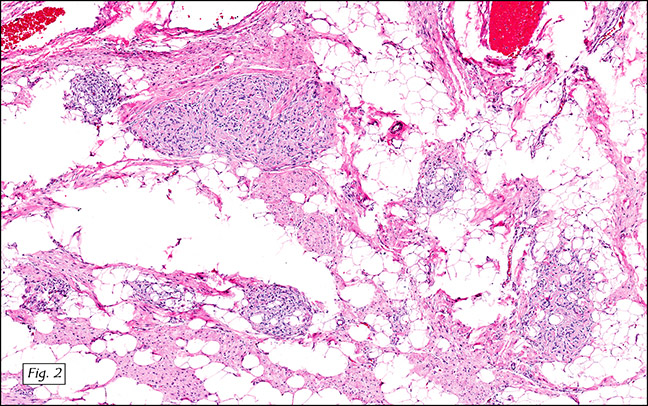
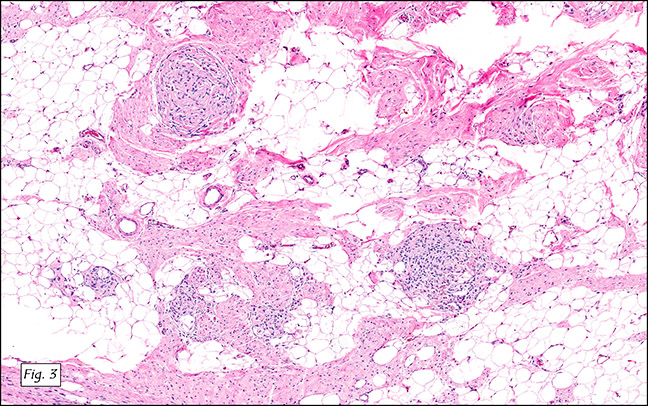
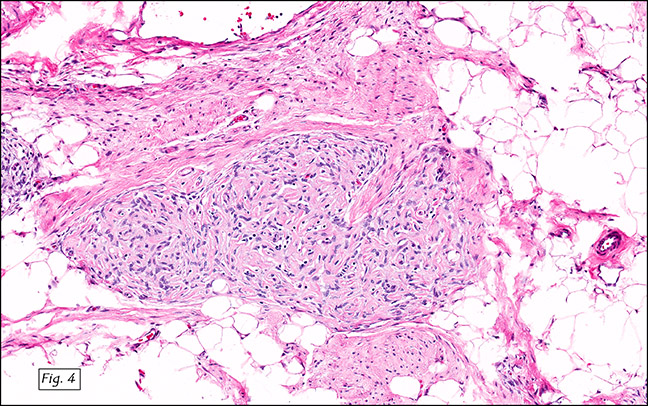
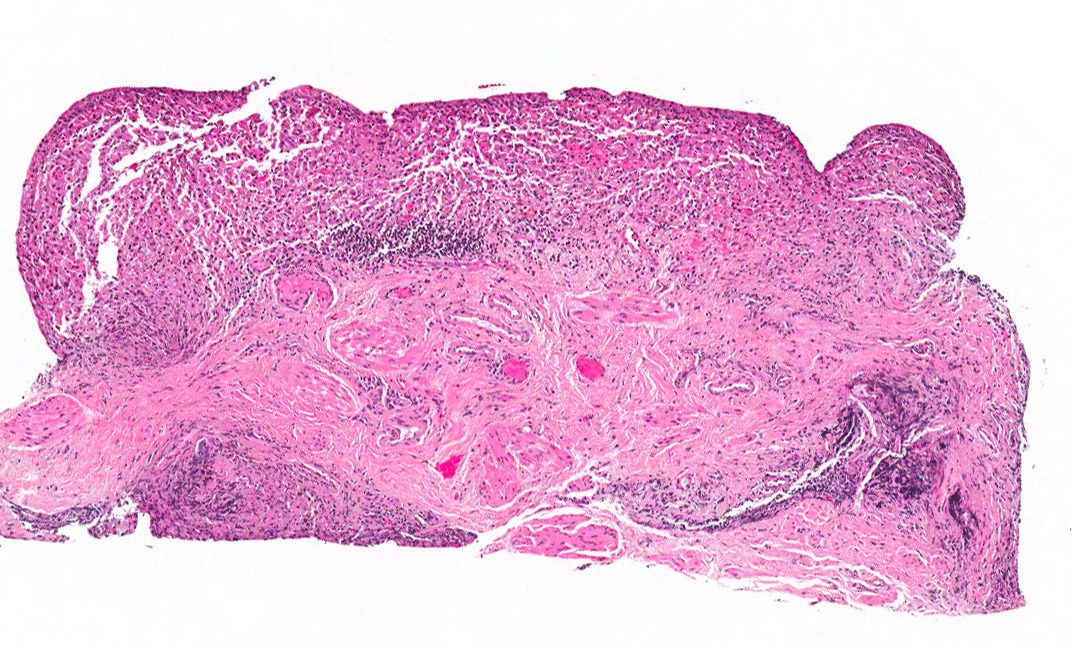
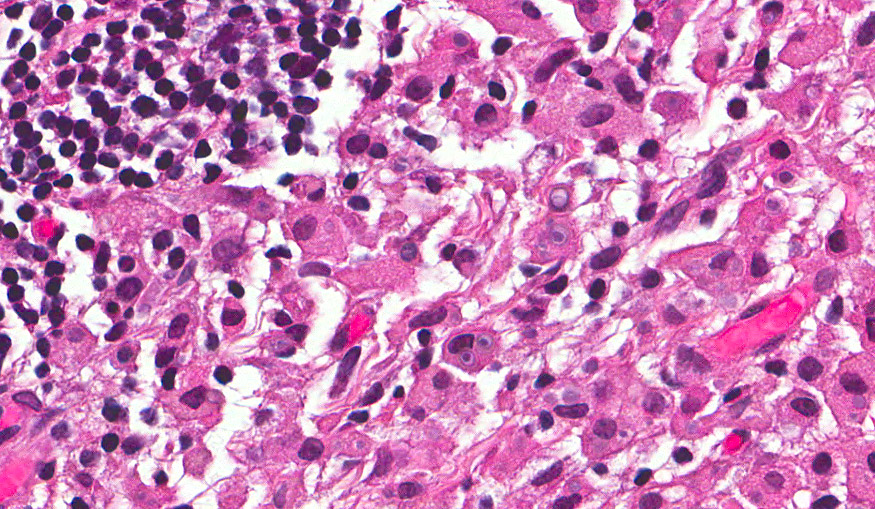
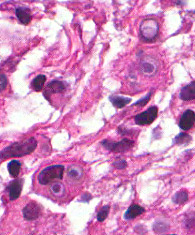
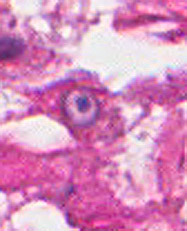
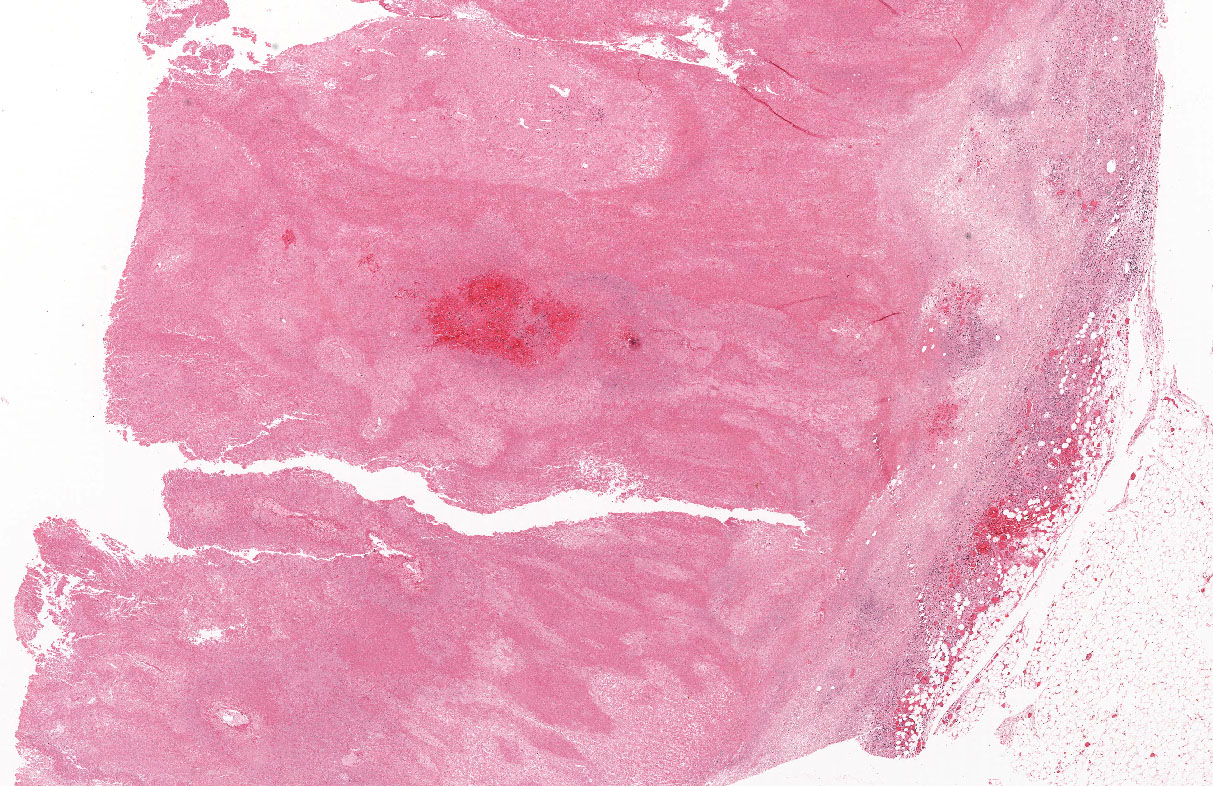
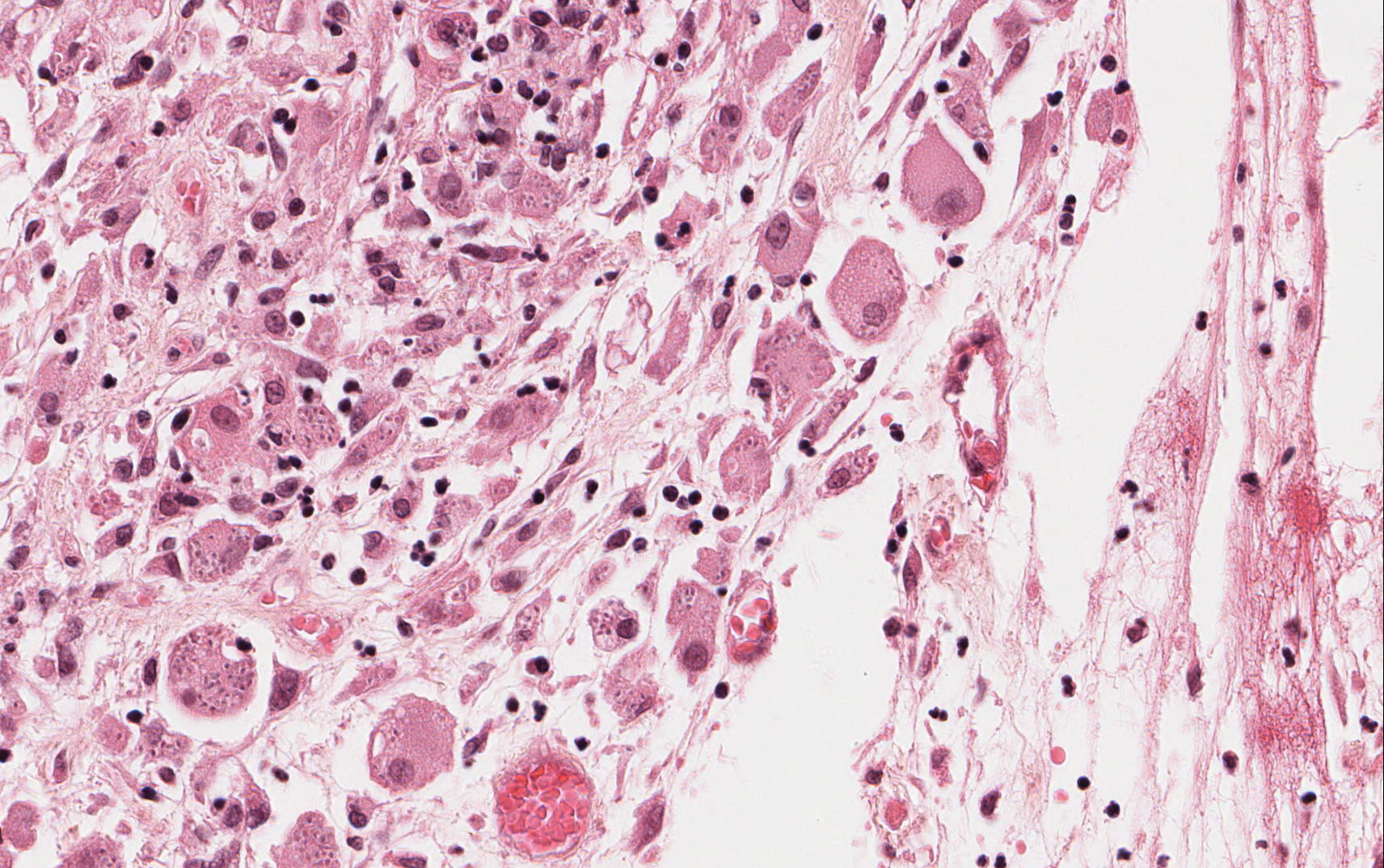
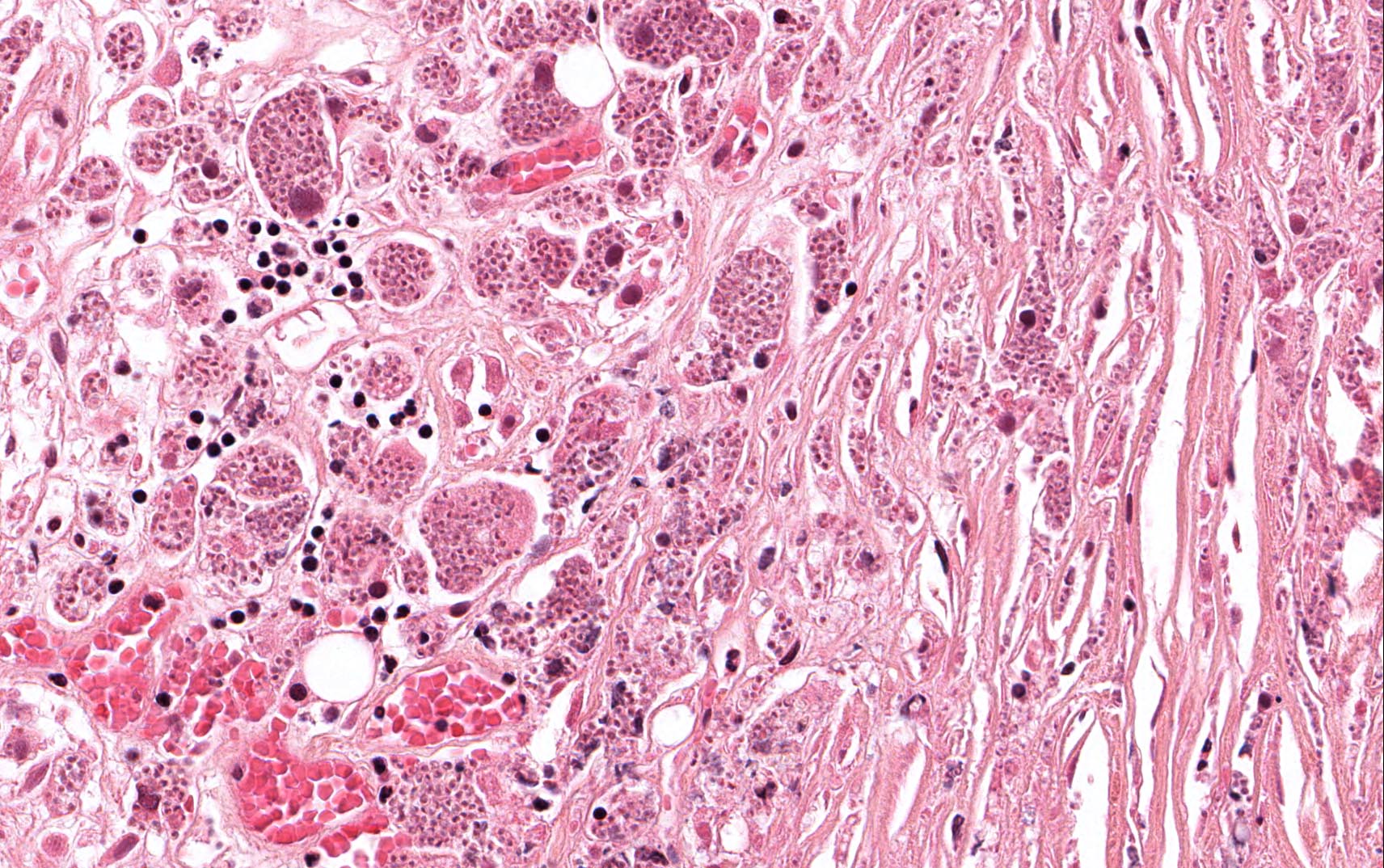
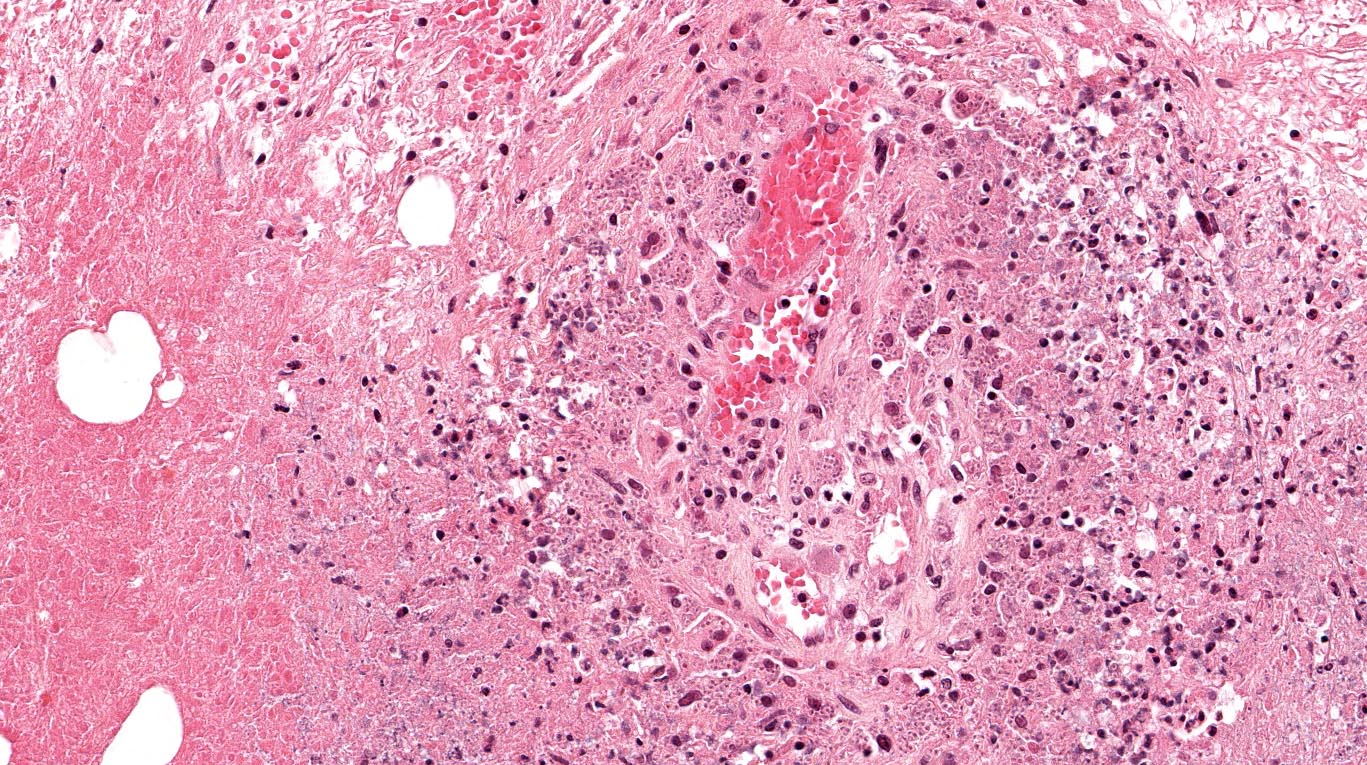
The excised specimen was 0.4 cm, tan-yellow and had urothelial denudation (Fig. 1). It involved the lamina propria, and to consisted of sheets of foamy histiocytes admixed with lymphocytes and plasma cells (Fig. 2). There were numerous small basophilic targetoid inclusions, most of which were within histiocytes, but some were extracellular (Figs 3a, 3b).
Diagnosis: Malakoplakia of Bladder
Li Lei M.D., PGY1 and Donald R. Chase, M. D.
Department of Pathology and Human Anatomy,
Loma Linda University Medical Center, Loma Linda, California
California Tumor Tissue Registry, Loma Linda, California
Discussion: Malakoplakia, or malacoplakia (MP) was first described by Michaelis and Gutmann in 1902. It was named for its gross features notably “soft†(“malako†in Greek) and plaque-like (“plakia†in Greek). Though it may be mistaken for a neoplasm clinically, the entity is actually an abnormal chronic granulomatous inflammatory process in response to infection.
Malakoplakia typically involves the genitourinary tract, particularly the bladder, followed by the gastrointestinal tract. Other sites that have rarely been reported include female genital tract, skin, tonsils, middle ears, larynx, lungs, brain, eyes, bone, thyroid, adrenal glands, retroperitoneum, liver, gallbladder, pancreas, and lymph node.
Clinically MP of the bladder shows a female predilection (4:1) with peak incidence in the fifth decade. It tends to present with hematuria, fever, and weight loss and many times is associated with recurrent urinary tract infection. Urine culture usually grows Escherichia coli or other bacteria. Findings on standard imaging are nonspecific. Both MRI and PET scans are helpful in the diagnosis. Magnetic resonance imaging (MRI) demonstrates nodules that are low in signal on T1, T2, early and late post gadolinium images with intervening fibrous stroma. Positron emission tomography (PET) scans show intense accumulation of fluoro-deoxyglucose.
Grossly, MP presents as solitary or multiple, soft, yellowish nodules or plaques involving the mucosa and underlying stroma. Lesions in bladder are often less than 2 centimeters, and are usually located in the region of the trigone. Focal necrosis is occasionally seen.
Microscopically, MP involves the lamina propria with sheets of epithelioid histiocytes with abundant granular eosinophilic cytoplasm, also known as von Hansemann cells. Lymphocytes, plasma cells and neutrophils are interspersed between the histiocytes. The pathognomonic finding is Michaelis-Gutmann bodies, which are basophilic targetoid or homogeneous inclusions, 3-10 micrometers in diameter. They are located in the cytoplasm or extracellularly in the stroma. Michaelis-Gutmann bodies can be highlighted by PAS-diastase stain, von Kossa stain for calcium, and Prussian blue stain for iron. Gram stain may show intracellular bacteria. Immunohistochemically, von Hansemann cells are positive for CD68, and negative for cytokeratin, calretinin, and Melan-A.
Electron microscopy shows Michaelis-Gutmann bodies to be concentrically layered structures with a central electron-dense core and radially oriented hydroxyapatite crystals. Von Hansemann cells have numerous phagolysosomes containing intact or partially digested bacteria, and lamellated crystalline bodies representing the early stage of Michaelis-Gutmann bodies.
The aforementioned ultrastructural findings are probably the most solid evidence we have so far to support bactericidal defect of macrophages as the etiology of malakoplakia. It has been proposed that impaired acidification of phagolysosomes results in bacterial accumulation within macrophages, which triggers the deposition of calcium, iron and phosphate, and then the formation of Michaelis-Gutmann bodies.
The pathogens are typically Gram-negative coliform bacteria, with Escherichia coli being the most common. In fact, a broad spectrum of bacteria has been involved, including other Gram-negative rods such as Proteus vulgaris and Pseudomonas as well as some Gram-positive cocci such as Staphylococcus aureus and Enterococcus. In patients with acquired immune deficiency syndrome (AIDS), the most common culprit is Rhodococcus equi, a Gram-positive coccobacillus. Mycobacterium, Candida albicans, human papillomavirus (HPV) and Epstein-Barr virus (EBV) are also rarely implicated, which sheds some light on the pathogenesis and supports the idea that malakoplakia is not necessarily bacteria-related.
To further complicate our understanding of malakoplakia, both lepromatous leprosy and Whipple’s disease are caused by macrophage dysfunction and subsequent intracellular accumulations of bacteria, but Michaelis-Gutmann bodies are not seen in either of them. These observations suggest that macrophage dysfunction followed by bacterial accumulation is not enough for malakoplakia to develop. Further research may help identify the unrecognized factor(s).
Overall, MP is a histologic diagnosis characterized by Michaelis-Gutmann bodies. The differential diagnosis, however, includes:
• Chemotherapy cystitis:
Cyclophosphamide, commonly used in the treatment of lymphoma, is notorious for causing hemorrhagic cystitis. Cystoscopic findings are mucosal edema and punctate hemorrhage other than soft plaques as seen in malakoplakia. Histologic findings include mucosal ulceration, vascular congestion, hemorrhage, and hemosiderin deposition.
• Xanthogranulomatous cystitis:
The characteristic finding is lipid-laden macrophages (“xanthoma cellsâ€) many times associated with multinucleated giant cells. Although it resembles MP, it lacks Michaelis-Gutmann bodies.
• Langerhans cell histiocytosis:
H&E staining reveals diffuse infiltrates of Langerhans cells, noted for their abundant foamy cytoplasm and indented bland nuclei. These cells are immunoreactive for CD1a and S-100. On electron microscopy, Birbeck granules are characteristic, but are absent in MP.
• Inflammatory pseudotumor:
The urine culture is characteristically negative. The mass is composed of spindled myofibroblasts and has a lymphoplasmacytic infiltrate. Immunostaining is positive for smooth muscle actin, desmin and anaplastic lymphoma kinase.
• Urothelial carcinoma:
Tumor cells show an infiltrative growth pattern, significant cytologic atypia, and immunoreactivities for cytokeratin 7 and 20. A prominent inflammatory background is not usually seen.
• Lymphoma:
Frank lymphoma is composed of predominantly CD20 positive, or rarely CD3 positive, tumor cells. However, early lymphoma may be mistaken for lymphocytic infiltrate in the background of malakoplakia.
Malakoplakia is most commonly seen in immunocompromised patients, such as those on immunosuppressive medications or chemotherapy, and patients with hematopoietic malignancies or diabetes mellitus. Radiation-induced local immunosuppression also poses a risk for malakoplakia, and these cases are easily confused with recurrent malignancies. Although MP by itself is non-neoplastic , it may be associated with various benign or malignant neoplasms, including lymphoma, adenomatous polyp, and carcinomas of the bladder, prostate, colon and endometrium. A case of renal malakoplakia followed by chronic myelomonocytic leukemia has recently been reported. Skinnider et al. reported a case of vaginal malakoplakia followed by diffuse large B-cell lymphoma localized to vagina. Retrospective review of the initial biopsy specimen with the aid of immunostaining showed that there had been malignant lymphocytes interspersed inconspicuously within the MP. A similar case of MP occurred in the bladder of a 63-year-old woman with a recurrent urinary tract infection. Eighteen months later she was found to have non-Hodgkin’s lymphoma localized to the bladder. Therefore, we believe that malakoplakia may serve as a potential red flag for future or concurrent disease processes.
Depending on the location and extent of disease, malakoplakia is treated by surgical excision and/or antibiotics that can reach high concentration in macrophages, such as ciprofloxacin, trimethoprim/sulphamethoxazol, and rifampicin. Cholinergic agonist bethanechol has been reported to improve lysosomal function by increasing intracellular cyclic guanosine monophosphate level, but the clinical effect is controversial. In patients on immunosuppressant therapy, adjustment of immunosuppressive medications may be necessary. With appropriate management, the prognosis of malakoplakia is usually excellent, though both spontaneous regression and fatal cases have been reported.
Suggested Reading:
Goldblum J, Folpe A, Weiss S. Enzinger & Weiss’ Soft Tissue Tumors, 6th ed: Philadelphia, Elsevier Inc, 2014; 374-6.
Rosai J. Rosai and Ackerman’s Surgical Pathology, 10th ed: Philadelphia, Elsevier Inc, 2011; 1251
Kradin RL1, Sheldon TA, Nielsen P, Selig M, Hunt J. Malacoplakia of the tongue complicating the site of irradiation for squamous cell carcinoma with review of the literature. Ann Diagn Pathol. 2012;16:214-8.
Batchelor JS1, Philp NH, Ramsden KL, Scott KW. Primary lymphoma of the bladder arising from an area of Malakoplakia. Br J Urol. 1991; 68:550-1.
Ngadiman S1, Hoda SA, Campbell WG, Gardner T, May M. Concurrent malakoplakia and primary squamous cell carcinoma arising in long-standing chronic cystitis. Br J Urol. 1994;74:801-2.
Abdou NI, NaPombejara C, Sagawa A, Ragland C, Stechschulte DJ, Nilsson U, Gousley W, Watanabe I, Lindsey NJ, Allen MS. Malakoplakia evidence of monocyte lysosome abnormality correctable by cholinergic agonist in vitro and in vivo. N Engl J Med. 1977;297:1413-9.
Biggar WD, Crawford L, Cardella C, Bear RA, Gladman D, Reynolds WJ. Malakoplakia and immunosuppressive therapy. Reversal of clinical and leukocyte abnormalities after withdrawal of prednisone and azathioprine. Am J Pathol. 1985;119:5-11.
Molnar JJ, Poliak A. Recurrent endometrial malakoplakia. Am J Clin Pathol. 1983;80:762-4.
Skinnider BF, Clement PB, MacPherson N, Gascoyne RD, Viswanatha DS. Primary non-Hodgkin’s lymphoma and malakoplakia of the vagina: a case report. Hum Pathol. 1999;30:871-4.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}