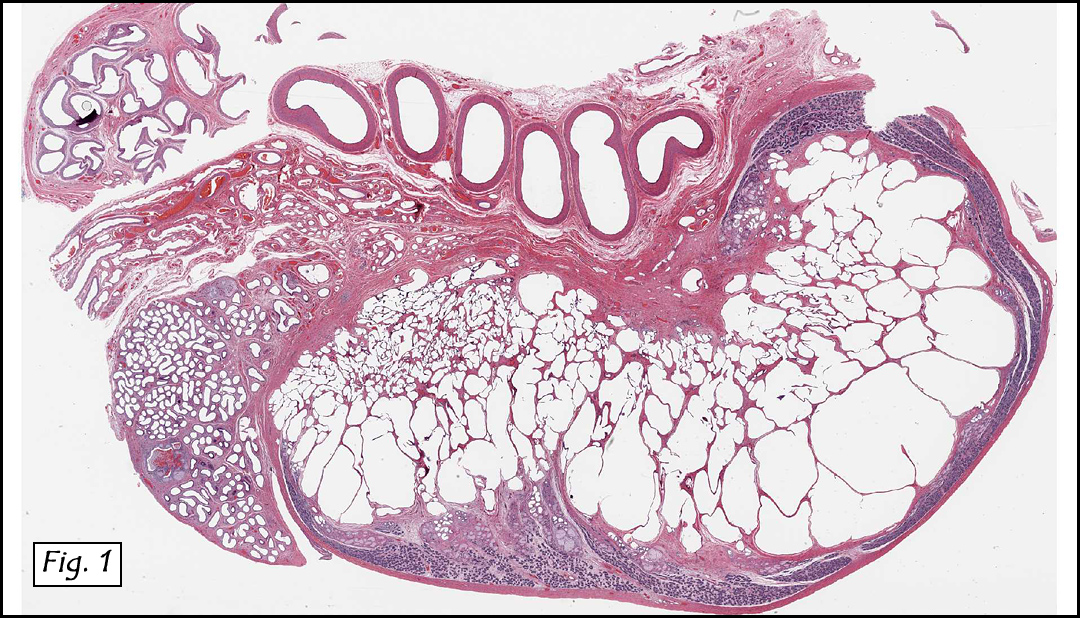
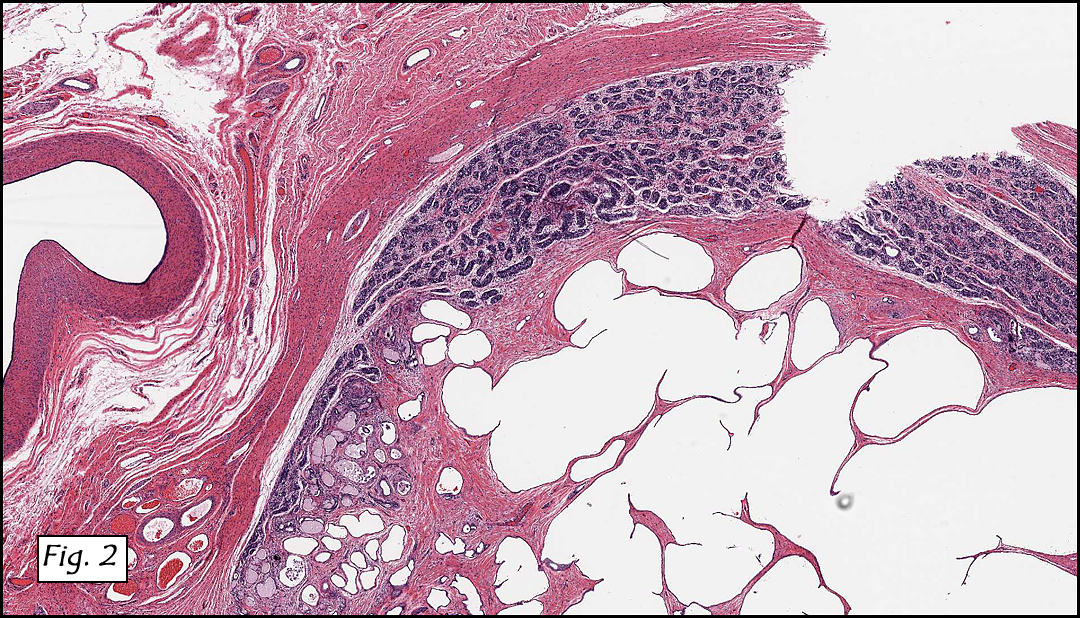
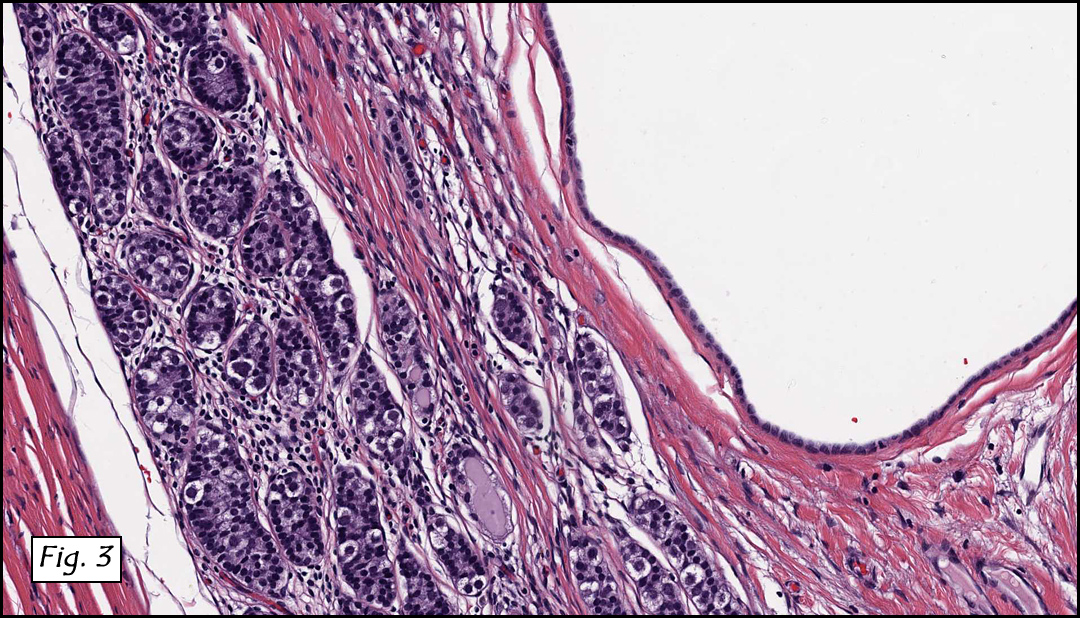
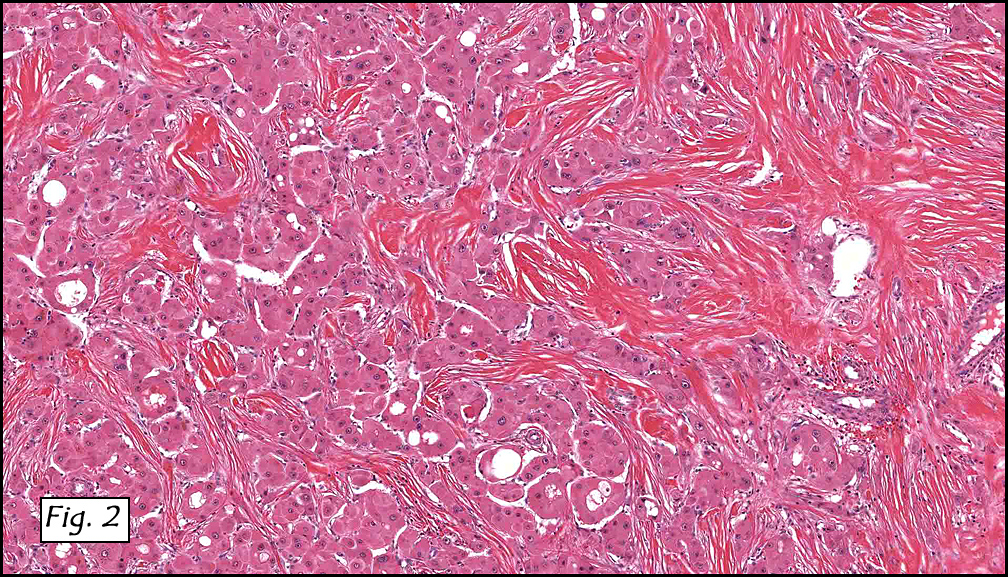
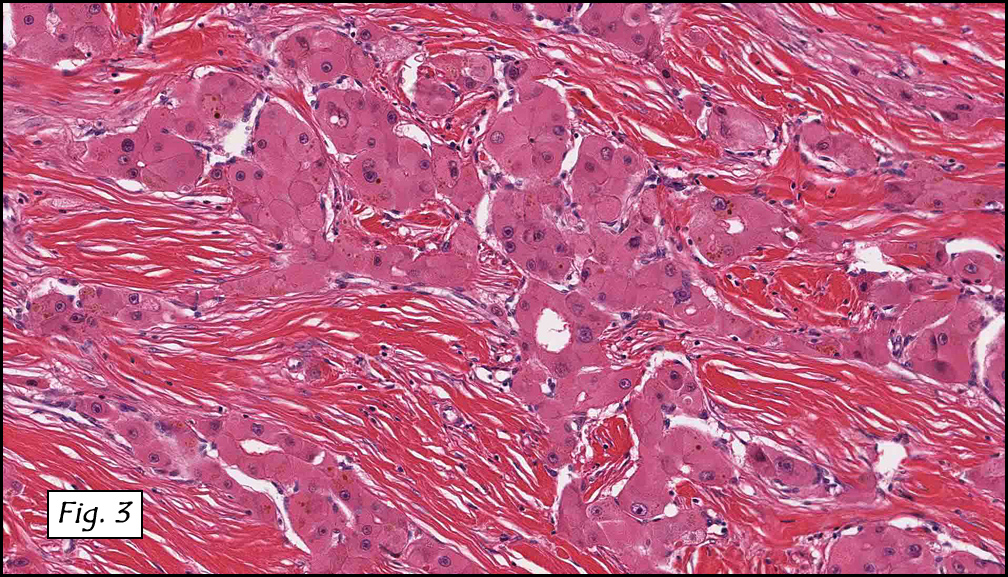
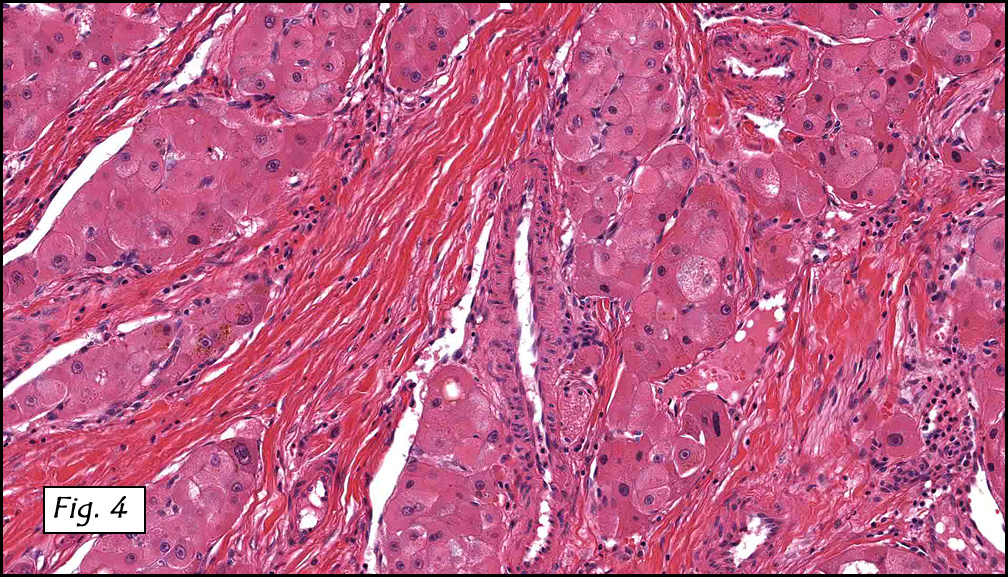
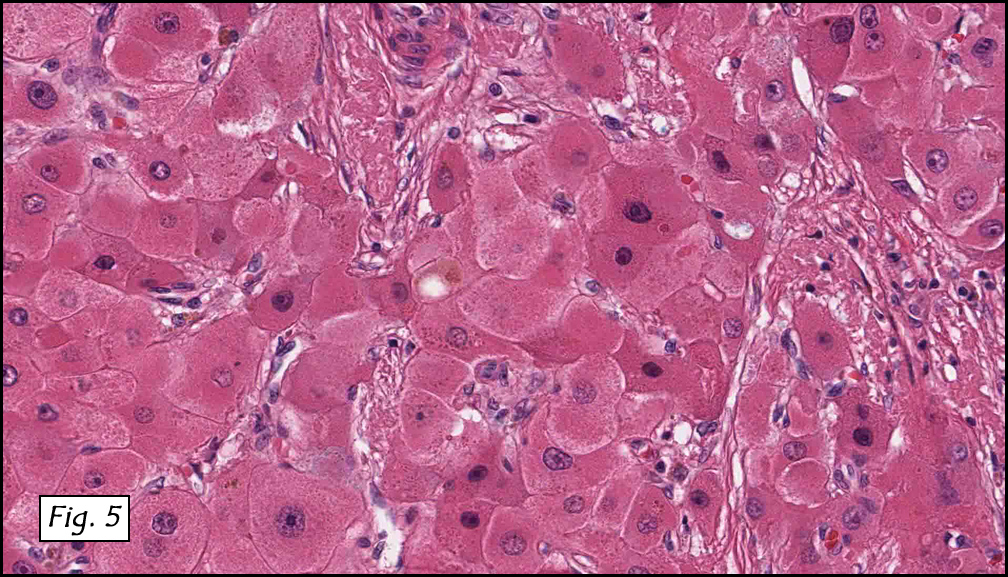
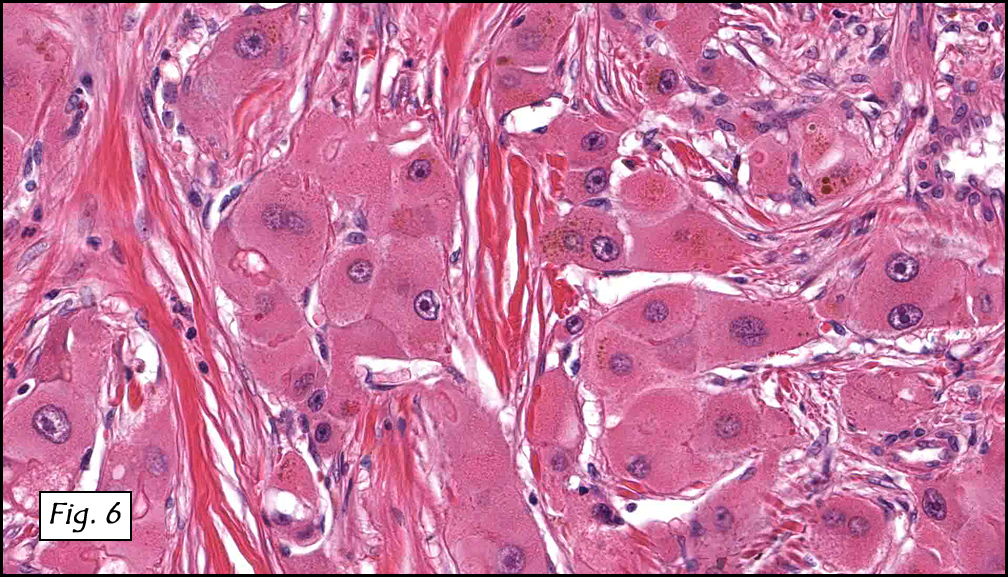
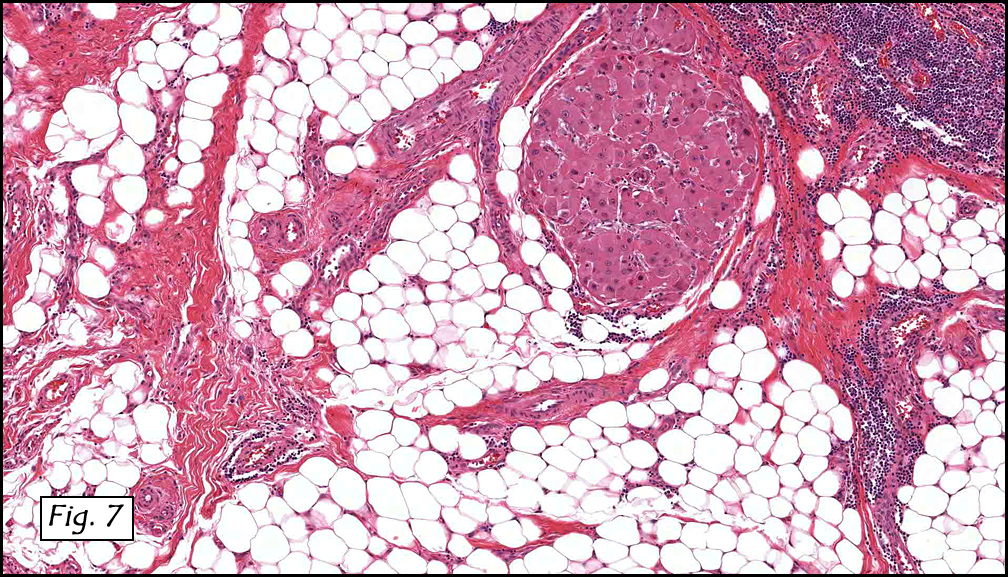
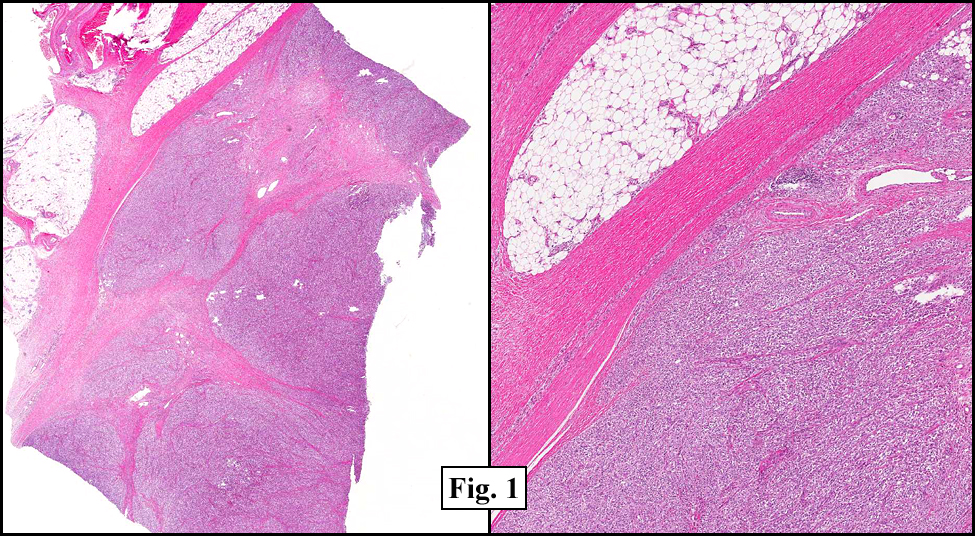
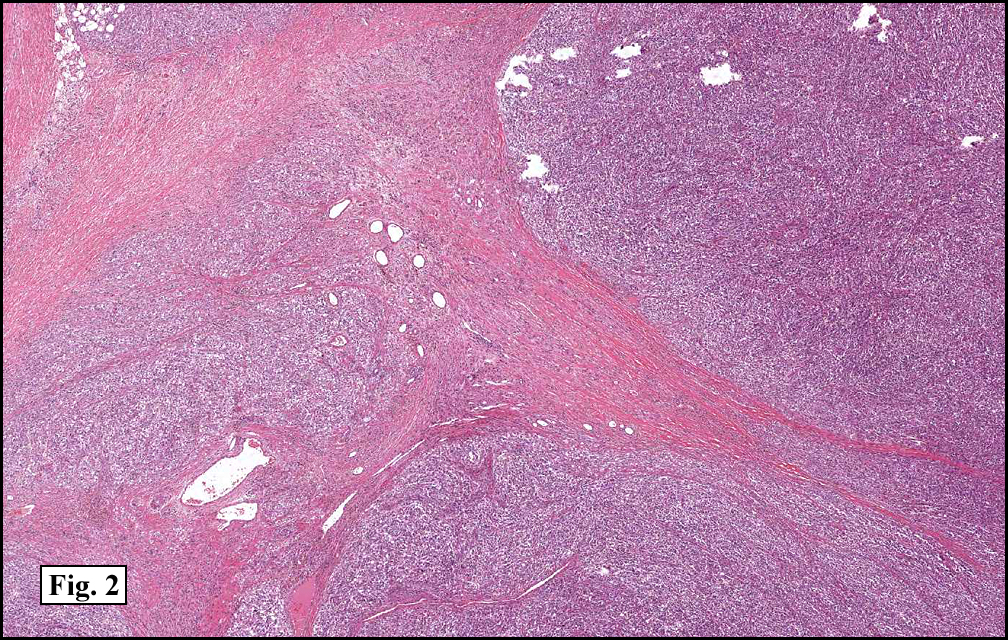
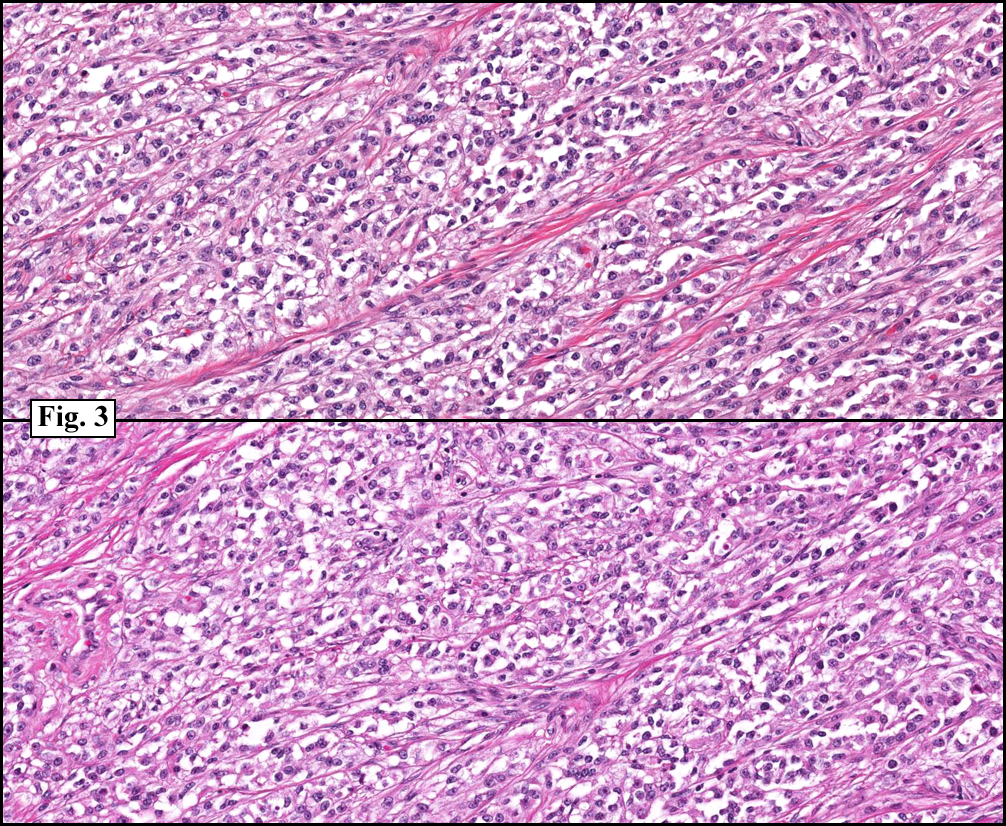
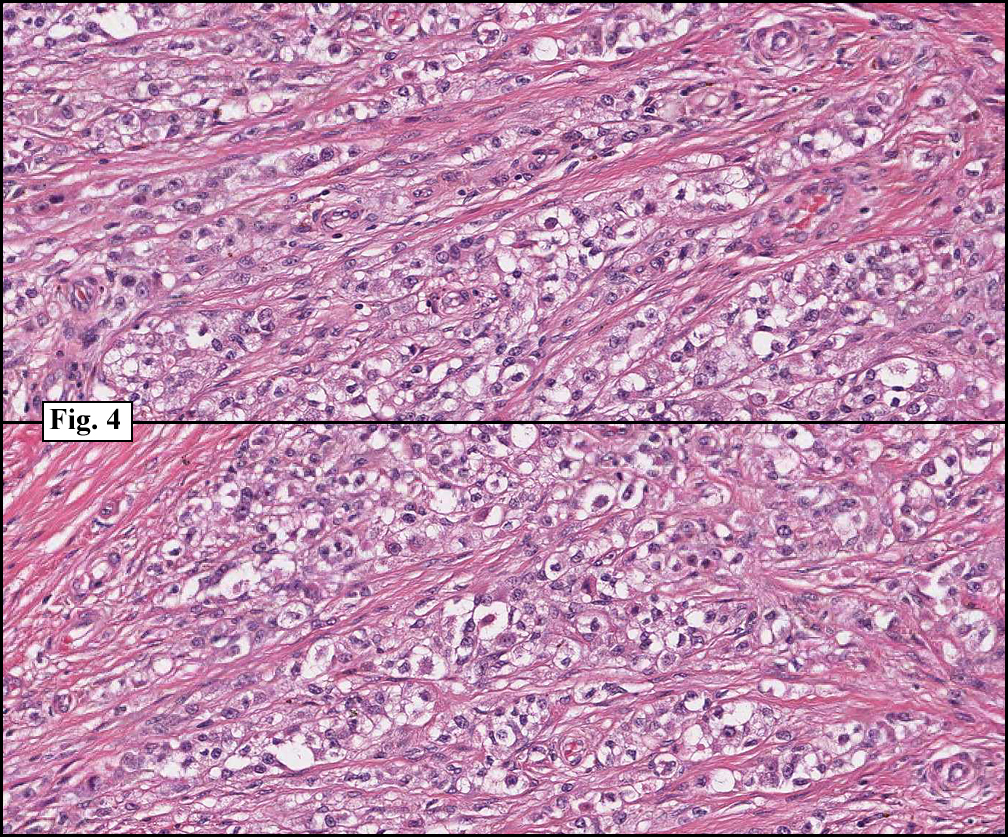
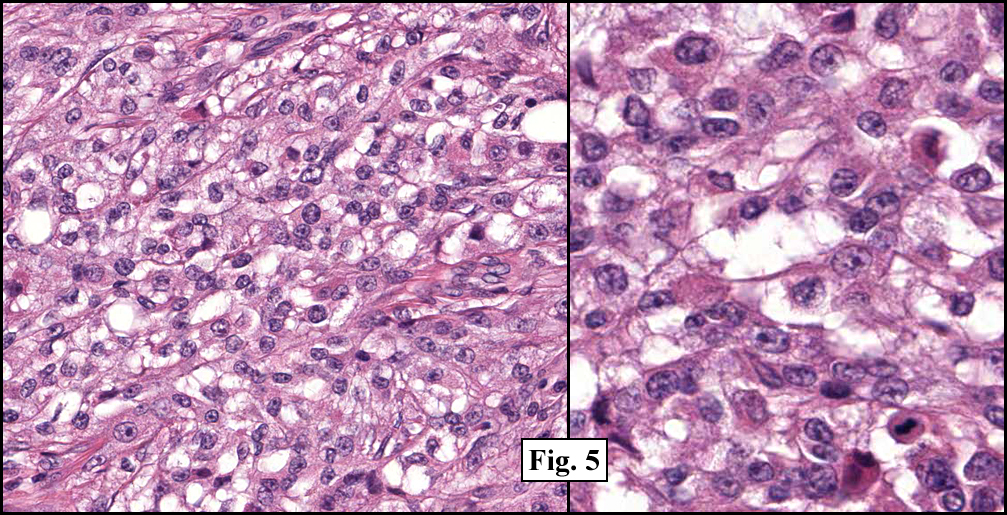
History: A three-year-old boy underwent orchiectomy for a cystic scrotal mass. Microscopically, the rete testis was found to be markedly distorted by cystic dilatation which compressed the adjacent seminiferous tubules (Figs. 1, 2). The cystic rete lining was of low cuboidal epithelium (Fig. 3).
{kind=link}
{kind=link}
{kind=link}
Diagnosis: Cystic dysplasia of the testis
Ellsworth BD, Zuppan C, Chase DR
Department of Pathology & California Tumor Tissue Registry
Loma Linda University Medical Center, Loma Linda, California
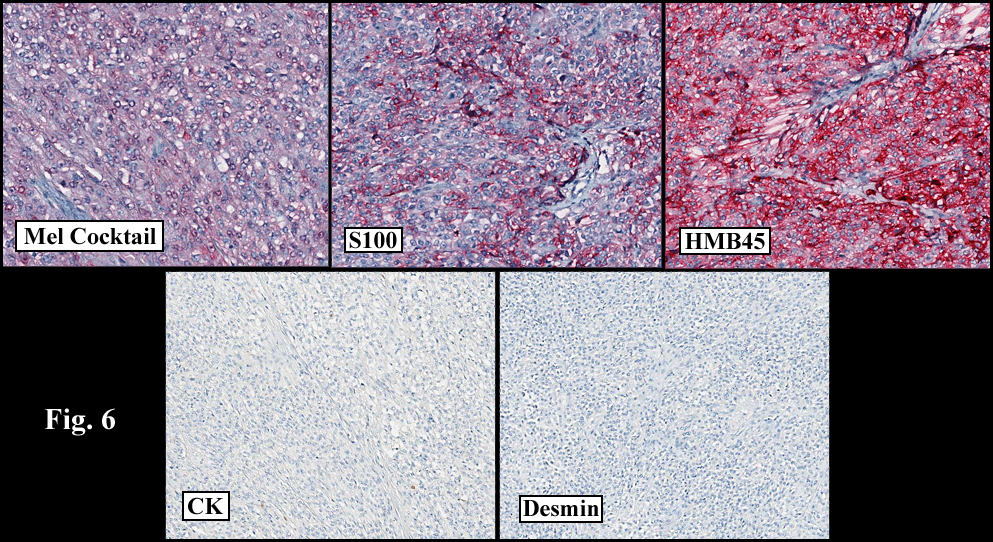
Discussion: Cystic dysplasia of the rete testis is a rare anomaly that usually presents in young boys as a unilateral painless scrotal mass. Approximately 50 cases have been documented. The cystic changes are well-appreciated by ultrasound and may be suspicious for malignancy. On ultrasound, variably-sized cysts are seen within the mediastinum testis with no changes in the testicular tissue and epididymis. Histologically, the rete testis is markedly dilated and cystic but lined by flat cuboidal epithelium. This process compresses and often obliterates the nearby testicular parenchyma which can render the testis infertile. Ipsilateral urinary tract anomalies are almost invariably present, most commonly renal agenesis or dysplastic kidney. For this reason, diagnosis of cystic dysplasia of the rete testis warrants further imaging of the urinary tract to look for additional anomalies.
The dysplastic changes are thought to occur as a result of improper development of the mesonephric duct. During normal embryogenesis, the mesonephric duct gives rise to the ureteral bud at its cephalic end which stimulates nephrogenesis of the metanephric blastema. From its caudal end, the extra testicular ducts (efferent ducts, epididymis, and vas deferens) derive. Defects in the mesonephric duct can result in anomalies in any of these structures. Normally, the rete testis and testicular cords (derived from the germinal epithelium) anastomose with the mesonephric-derived extra testicular ducts at the efferent ductules. Failure of this process causes anomalies and results in cystic distortion of the rete testis. With this relationship in mind, Nistal et al have theorized that inappropriate (pre-pubertal) secretory activity by the rete testis and fluid entrapment are responsible forecyst development, a process that occurs until puberty when the seminiferous tubules canalize and develop lumens allowing the fluid to relocate. This process seems to have occurred in two reported cases of resolved cystic dysplasia. In both cases, resolution was relatively sudden and occurred near the time of (reference – Thomas 2003).
Treatment commonly consists of unilateral orchiectomy. Testis-sparing enucleation is occasionally performed to spare hormonally active tissue, but yields a chance of recurrence. As no malignant behavior has been observed and two reported cases have regressed, conservative therapy or watchful waiting has been suggested.
Suggested Reading:
Keetch DW, McAlister WH, Manley CB, Dehner LP. Cystic dysplasia of the testis. Pediatr Radiol. 1991;21:501-503.
Nistal M, Regadera J, Paniagua R. Cystic dysplasia of the testis. Light and electron microscopic study of three cases. Arch Pathol Lab Med.1984;108:579-583.
Thomas AD, Wu H, Canning DA, et al. Spontaneous regression of cystic dysplasia of the testis. J Urol. 2003;169:645.
Wojcik LJ, Hansen K, Diamond DA, et al. Cystic dysplasia of the rete testis: a benign congenital lesion associated with ipsilateral urological anomalies. J Urol. 1997;158:600-604.
Zaragoza M, Buckler L, Parikh M. Cystic dysplasia of the testis: An unusual cause of a pediatric scrotal mass. Urology. 1996;47:244-247.
Robson W, Thomason M, Minette L. Cystic dysplasia of the testis associated with multicystic dysplasia of the kidney. Urology. 1998;51:477-479.
Camassei F, Francalanci P, Ferro F, et al. Cystic dysplasia of the rete testis: Report of two cases and review of the literature. Pediatr Dev Pathol. 2002;5:206-210.
Erberli D, Gretener H, Dommann-Scherrer C, et al. Cystic dysplasia of the testis: A very rare pediatric tumor of the testis. Urol Int.2002;69:1-6.
Fisher JE, Jewett Jr TC, Nelson SJ, et al. Ectasia of the rete testis with ipsilateral renal agenesis. J Urol 1982;128:1040-3.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}