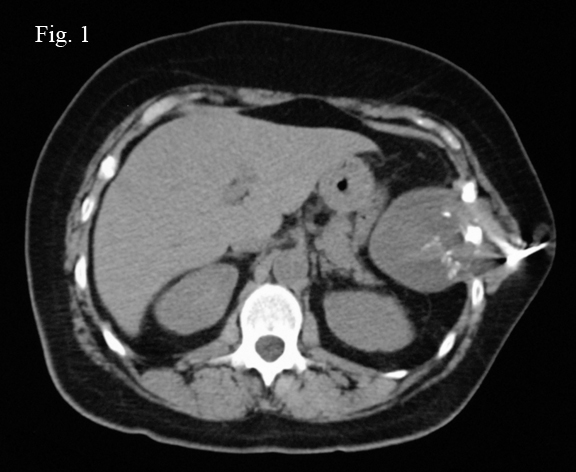
History: A 50 year-old woman presented with a 9 x 7 x 6 cm slowly enlarging, painless upper abdominal mass extending to the flank. A CT scan demonstrated a focally calcified, well-demarcated tumor which completely encompassed and obliterated one rib with involvement of adjacent ribs, as well as extending peripherally into the soft tissues of the thoracic wall (Fig. 1). She subsequently underwent needle biopsy resulting in a small aggregate of gray-tan semi-translucent tissue fragments.
{kind=link}
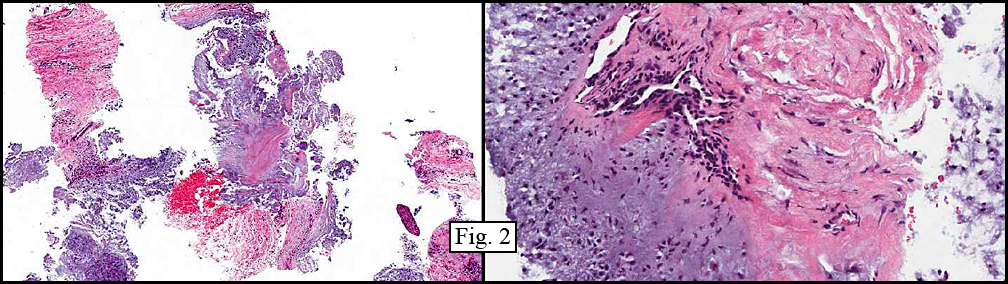
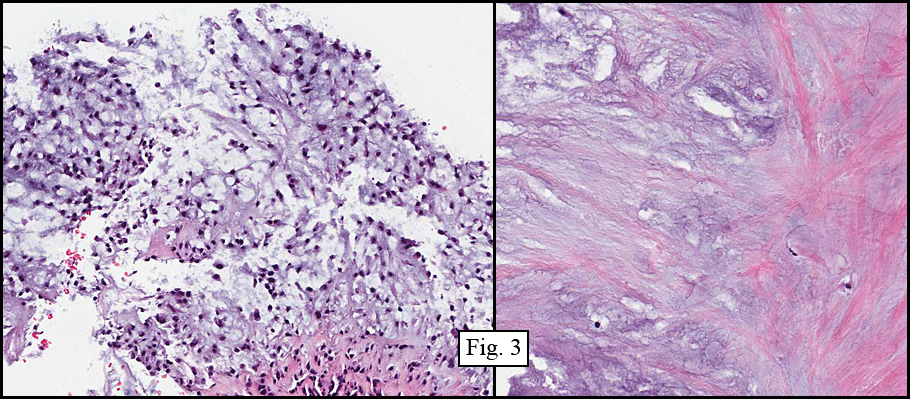
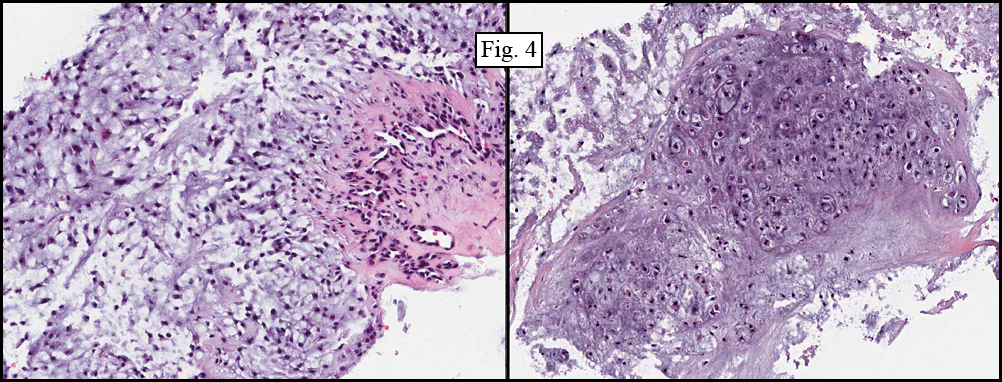
Microscopically, the material varied greatly in cellularity, including fibrous regions, prominent supporting blood vessels, and abundant myxomatous regions incorporating bland round to oval cells with occasional stellate cells (Fig. 2). Mitotic figures were not encountered. The predominant pattern was myxomatous and had no particular growth pattern (Fig. 3 left). There were also regions of necrosis and fibrosis in which were scattered hyphal mycotic organisms (Fig. 3 right). In addition to haphazard growth (Fig. 4 left), several areas showed nodular patterns with groupings of small, hyperchromatic cells residing in lacunae (Fig. 4 right.). Physaliferous cells were not encountered.
{kind=link}
{kind=link}
{kind=link}
Diagnosis: “Myxoid Chondrosarcomaâ€
Laura Denham MS3, Donald R. Chase, M.D.
Department of Pathology, Loma Linda University and Medical Center
California Tumor Tissue Registry, Loma Linda, California
Discussion: Myxoid Chondrosarcoma (MC) is a rare soft tissue tumor that comprises less than three percent of all soft tissue tumors. It was first described by Stout and Verner in 1953 but not until 1972 did Enzinger and Shiraki elaborate on the clinicopathologic features. MCs have traditionally been divided into skeletal and extraskeletal tumors based on their location of origin, which at times may be difficult to elucidate. It is believed that extraskeletal myxoid chondrosarcoma is a distinct entity whereas skeletal myxoid chondrosarcoma is a separate variant of conventional chondrosarcoma. In 1985 it was discovered that the majority of extraskeletal myxoid chondrosarcomas showed a genetic translocation which was lacking in neoplasms arising from bone. This finding, along with certain histologic and clinical findings, suggests that they are separate entities.
The tumor is grossly characterized as being well circumscribed, lobulated to nodular, and having a gelatinous gray to tan cut surface. There is often hemorrhage, which may be quite prominent. Microscopically, it is usually composed of fairly uniform small round to oval cells with hyperchromatic nuclei edged by eosinophilic cytoplasm. The cells are often arranged in cords, strands and/or nests in a myxoid matrix which may be separated into lobules by fibrous tissue. Occasionally the cells may be arranged in a more diffuse pattern, which is reportedly more common in the skeletal version of myxoid chondrosarcoma. Although somewhat unusual, it is possible to find foci of well-differentiated chondrocytes with typical lacunae in a specimen that has been well sampled, however calcification is rare. Mitoses and fibroblastic cells may also occasionally be found. A cellular variant of myxoid chondrosarcoma is described, characterized by sheets of large cells with prominent nucleoli and a more open, vesicular chromatin pattern and with less myxoid change between cells. Rarely, areas of blue cells reminiscent of Ewings sarcoma/PNET, as well as rhabdoid-like cells with peri-nuclear inclusions have been reported.
Neoplastic cells stain positively for S100 protein in 20-40% of the cases. The decoration is generally weak, but diffuse. Because many of the differential diagnoses also stain strongly for S100 protein, Enzinger and Weiss argue that the usual lack of staining in MC is a useful diagnostic feature. Curiously, MCs occasionally stain positively for cytokeratins, such as EMA and may also stain positively for chromogranin and neuron-specific enolase, suggesting a possible neuroendocrine lineage. Neurosecretory granules seen via electron microscopy also lend support this possibility. Other ultrastructual and histochemical findings such as dilated rough endoplasmic reticulum, prominent mitochondria, diastase sensitive PAS positive intracytoplasmic material and hyaluronidase resistance tend to support chondroid lineage.
Genetic studies show approximately 70% of extraskeletal myxoid chondrosarcomas have a t(9; 22) (q22; q12) translocation. This balanced translocation combines the EWS gene with the CHN (known as NOR1 in rodents). Other translocations have also been described, however, to date similar translocations have not been found in the skeletal myxoid chondrosarcoma.
Differential Diagnosis:
• Chondromyxoid fibroma – Typically the tumor cells are arranged in fused lobules, with a fibrous band surrounding the lesion. The periphery of a lobule tends to be more cellular, with more pleomorphic and fibroblastic cells, while the center of the lobule is usually more myxomatous and may show lacunae formation. It is not unusual to find multinucleated giant cells, especially in areas with bland cells. Occasional foci of calcification or ossification may also be present. An area of well-vascularized connective tissue separates lobules from each other, as well the neoplasm from adjacent bone.
• Ossifying Fibromyxoid Tumor of Soft Parts (OFMT) – Grossly the tumor is surrounded by a thick fibrous capsule and is separated into lobules by fibrous tissue with an underlying shell of lamellar bone in the majority of cases. The uniform round to spindled cells are rimmed by pale cytoplasm and are found in nests, cords or sheets within a myxoid to collagenous matrix. There may be areas of cartilage formation, as well as intricate vasculature often with perivascular hyalinization, both of which are rare in myxoid chondrosarcomas. Furthermore, the majority of OFMTs are S100 positive.
• Myxoid liposarcoma – Grossly these tumors are gelatinous and may be multinodular. The round to spindled cells are characteristically denser around the periphery and are suspended in a myxoid matrix, which is sensitive to hyaluronidase. Lipoblasts are normally present in varying numbers, usually at the periphery of a tumor lobule. A distinct plexiform “chicken-wire†vasculature is a common feature, which helps to differentiate this tumor from myxoid chondrosarcomas. There may also be foci of high cellularity which may or may not have round cell differentiation and little myxoid stroma.
• Myoepithelioma – This mixed tumor may be composed of a mixture of epithelioid, clear, spindled or physaliferous cells found in cords, nests or sheets. The stromal component is also variable and may be myxoid, chondroid or focally osteoid. If ductal differentiation is not present, it may be difficult to differentiate between this entity and myxoid chondrosarcoma. However, in contrast to myxoid chondrosarcomas, these tumors typically co-express S100 and epithelial markers such as EMA and cytokeratin, and may express smooth muscle actin as well.
• Myxoma – A fairly common neoplasm, myxomas are essentially avascular, consisting of widely-spaced, bland spindled to stellate cells with round to oval nuclei in an abundant myxoid matrix with varying amounts of reticulin fibers. There may also be infiltration of the neoplasm into the surrounding skeletal muscle. Myxomas are much less cellular than myxoid chondrosarcomas and tend to have a haphazard spindled pattern rather than a lobular chondrocyte-type pattern.
• Myxoid variant of Extraskeletal (Soft Part) Chondroma – These multinodular lesions are composed of chondrocytes, which tend to be more differentiated toward the periphery of the lesion. The cells may be surrounded by stippled calcification and may occasionally have foci of multinucleated giant cells. Although they are usually less cellular than myxoid chondrosarcoma, the two entities may be difficult to differentiate. However, myxochondromas may be distinguished from myxoid chondrosarcomas based on their predilection for hands and feet, their propensity to form mature hyaline cartilage, and their unique peri-cellular granular calcification pattern.
• Chondroid Lipoma – This rare tumor is generally smooth, well circumscribed and consists of hibernoma-like cells with vacuolated, often granular cytoplasm. The cells are in cords or nests with surrounding myxoid stroma. Although some cells may resemble chondroblasts, this benign neoplasm can be distinguished from myxoid chondrosarcomas by the intracytoplasmic vacuoles, rounded, less lobular appearance, greater vascularity and more consistent positive staining of S100 protein.
Clinically, myxoid chondrosarcomas are mostly found in the deep tissues of the lower extremities, especially buttock and thighs, as well as in the proximal upper extremities such as shoulders or neck. Both skeletal and extraskeletal forms of MC are characterized by slow growth. However, despite its bland morphology and slow growth pattern extraskeletal myxoid chondrosarcoma has a propensity for metastases, most commonly to the lung. Furthermore, recurrences are very common in this entity and may often be multiple. Both chemotherapy and radiation have little role in the eradication of disease, and early total surgical extirpation is the preferred treatment.
Suggested Reading:
1. Enzinger FM, Skiraki M. Extraskeletal myxoid chondrosarcoma: an analysis of 34 cases. Hum Pathol. 3:421-435, 1972.
2. Meis-Kindblom JM, Bergh P, Gunterberg B, Kindblom LG. Extraskeletal myxoid chondrosarcoma: a reappraisal of its morphologic spectrum and prognostic factors based on 117 cases. Am J Surg Pathol 23:636-50, 1999.
3. Atonescu CR, Argani P, Erlandson RA, healey JH, Ladanyi M, Huvos AG. Skeletal and extraskeletal myxoid chondrosarcoma: a comparative clinicopathologic, ultrastructural and molecular study. Cancer 83:1504-21, 1998.
4. Rubin BP, Fletcher JA. Skeletal and extraskeletal myxoid chondrosarcoma: related or distinct tumors? Adv Anat Pathol 6:204-212, 1999.
5. Meis JM, Enzinger FM. Chondroid lipoma: a unique tumor simulating liposarcoma and myxoid chondrosarcoma. Am J Surg Pathol 17(11): 1103-1112, 1993.
6. Rahimi A, Beabout JW, Ivins JC, Dahlin DC. Chondromyxoid fibroma: a clinicopathologic study of 76 cases. Cancer 30:726-736, 1972.
7. Kilpatrick SE, Hitchcock MG, Kraus MD, Calonje, E, Fletcher C. Mixed tumors and myoepitheliomas of soft tissue: a clinicopathologic study of 19 cases with a unifying concept. Am J Surg Pathol 21(1):13-22, 1997.
8. Mackenzie DH. The myxoid tumors of somatic soft tissues. Am J Surg Pathol 5(5): 443-458, 1981.
9. Smith TA, Easley KA, Goldblum JR. Am J Surg Pathol 20(20):171-180, 1996.
10. Enzinger FM, Weiss SW, Liang CY. Ossifying fibromyxoid tumor of soft parts: a clinicopathological analysis of 59 cases. Am J Surg Pathol 13(10) 817-827, 1989.
11. Enzinger and Weiss, Soft Tissue Tumors, 5th edition.