History: A 36 year old female presented with pelvic pain. An abdominal ultrasound was performed and identified a mass on the round ligament.
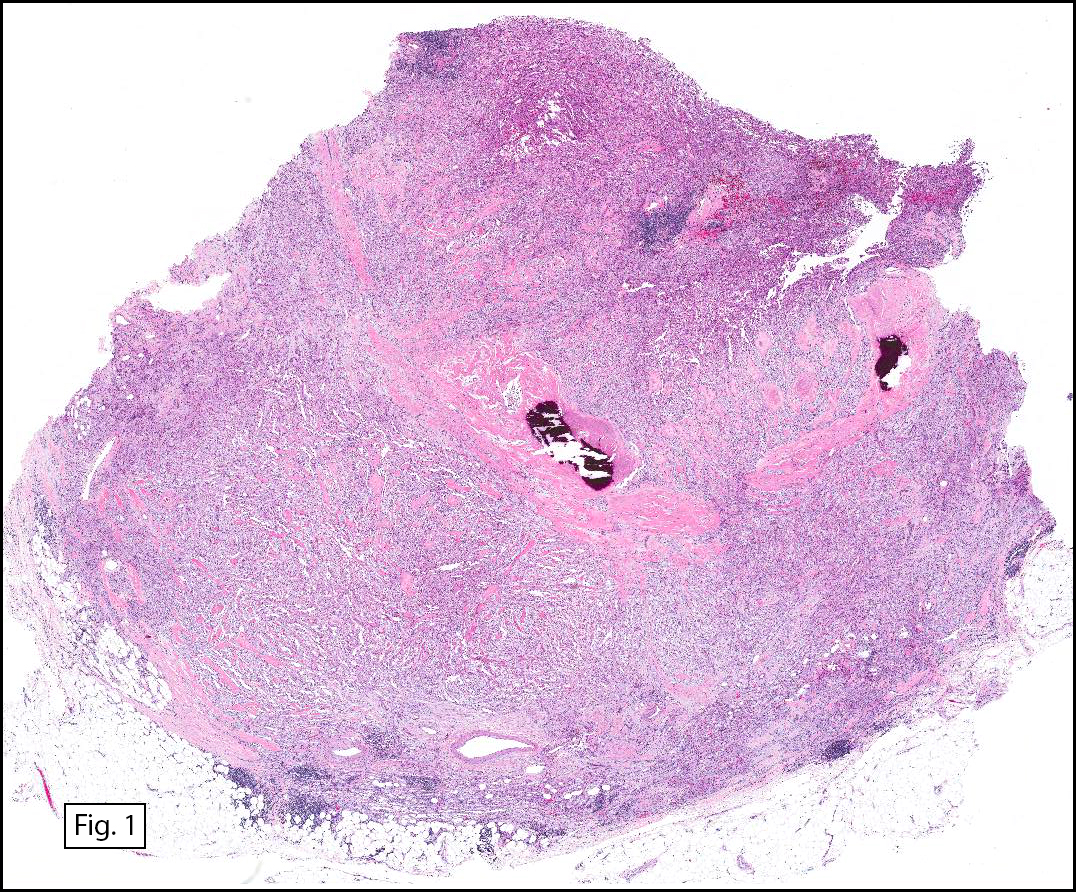
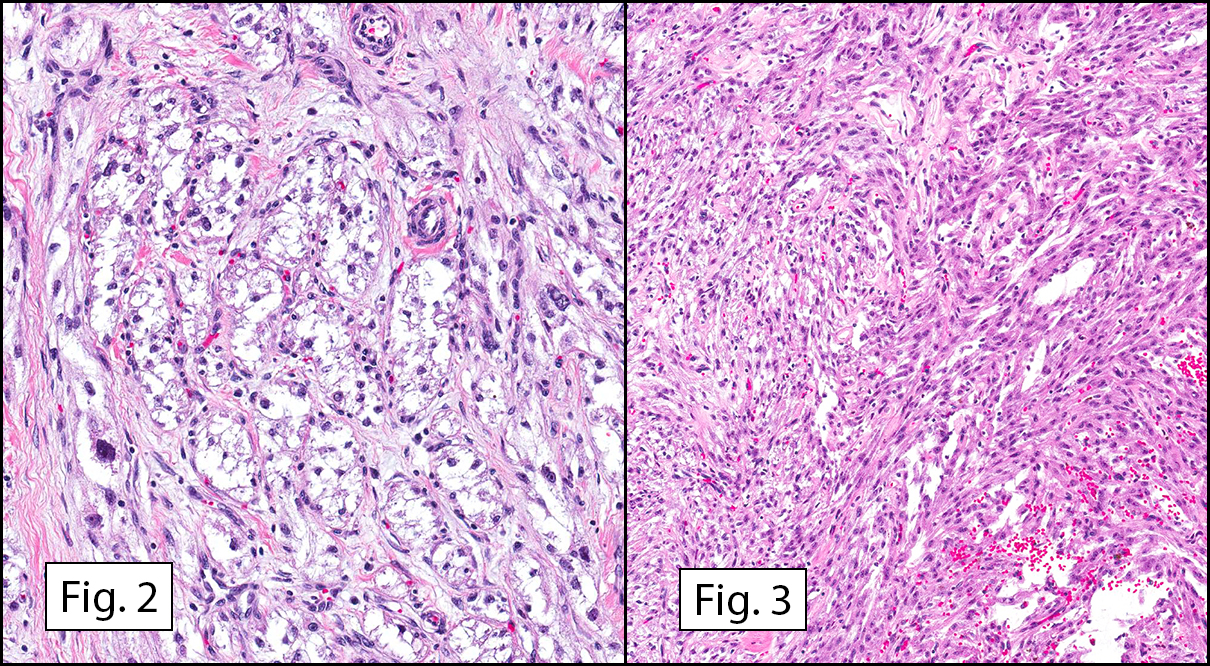
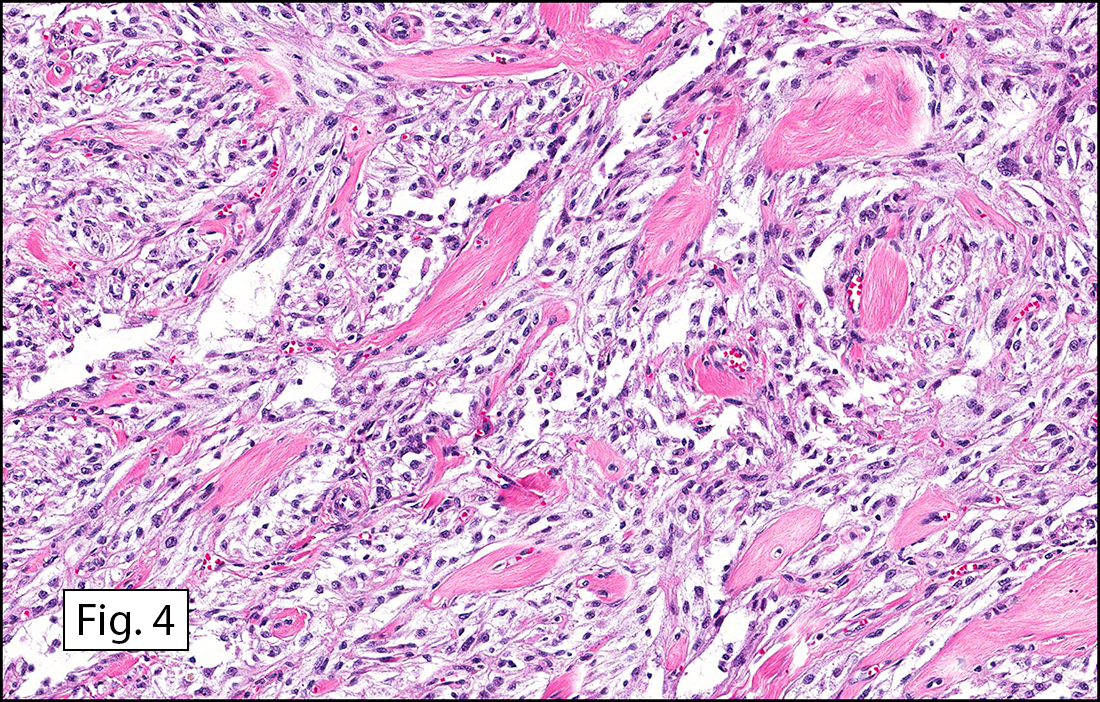
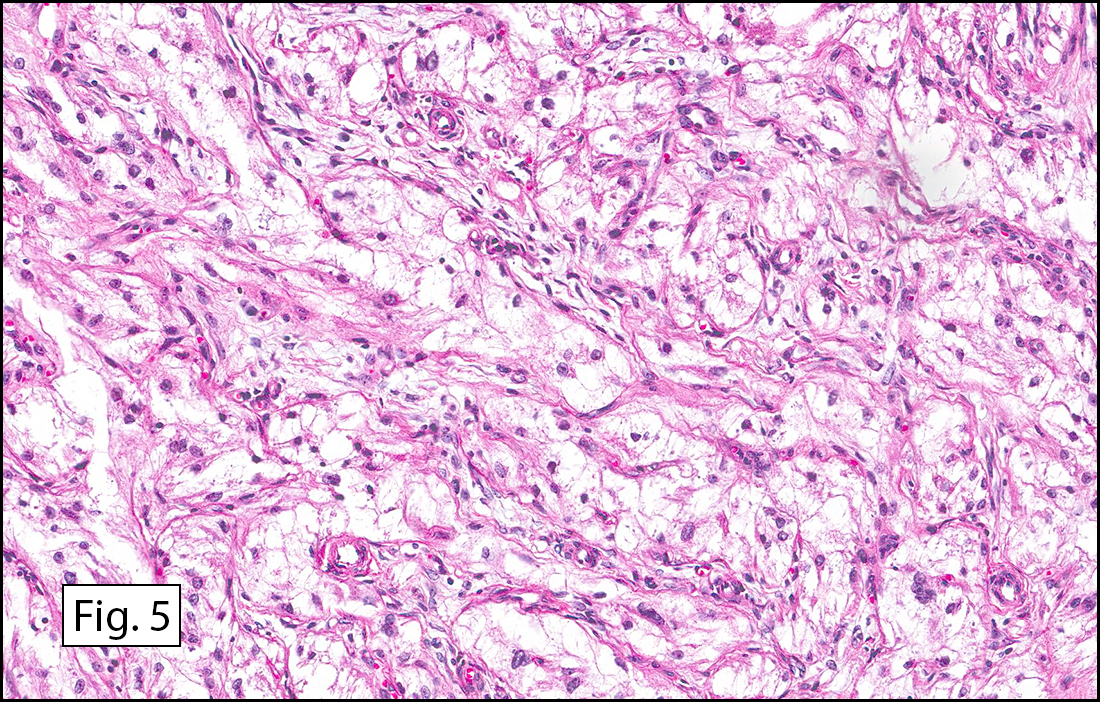
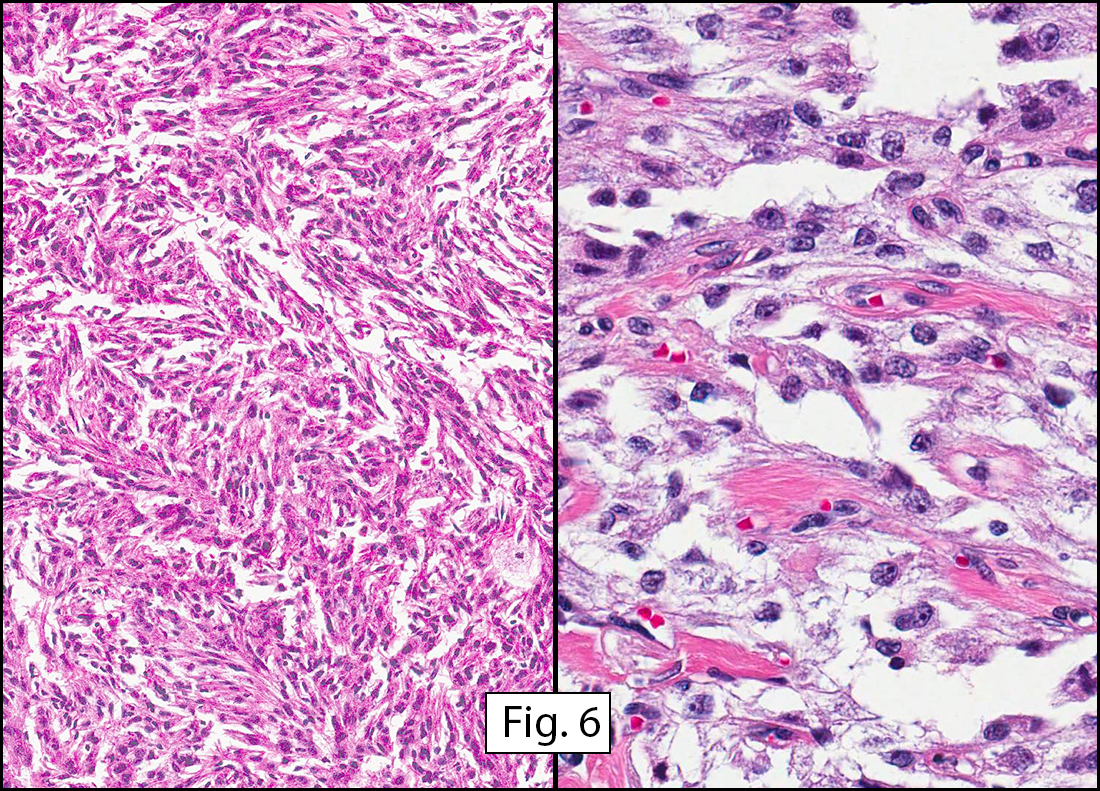
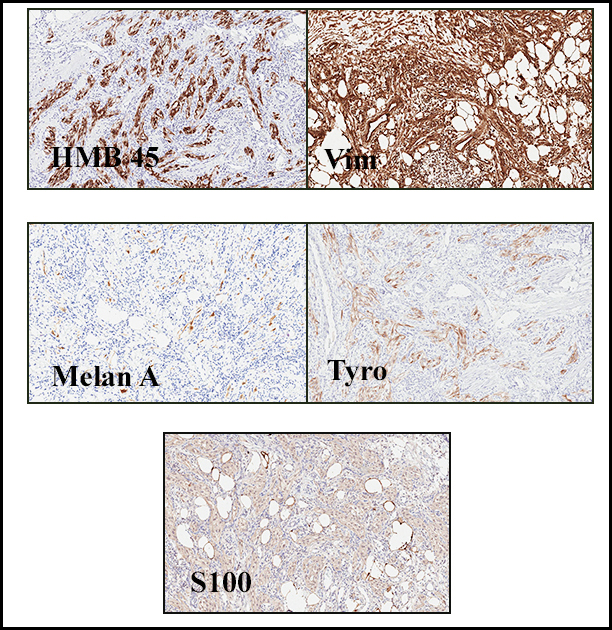
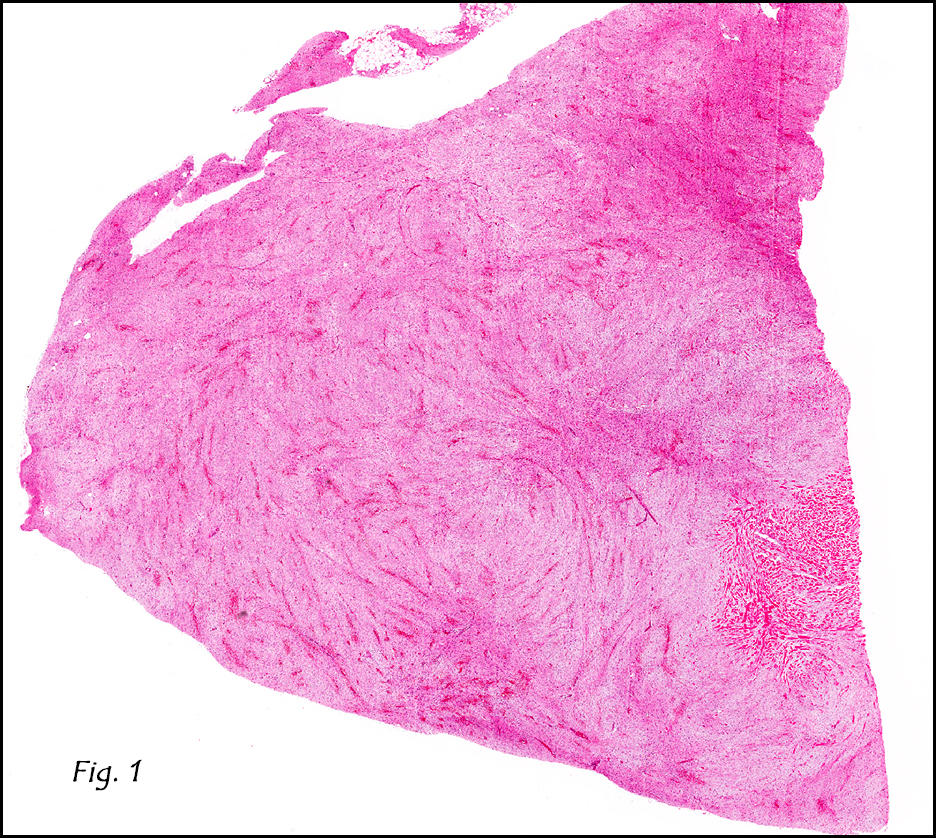
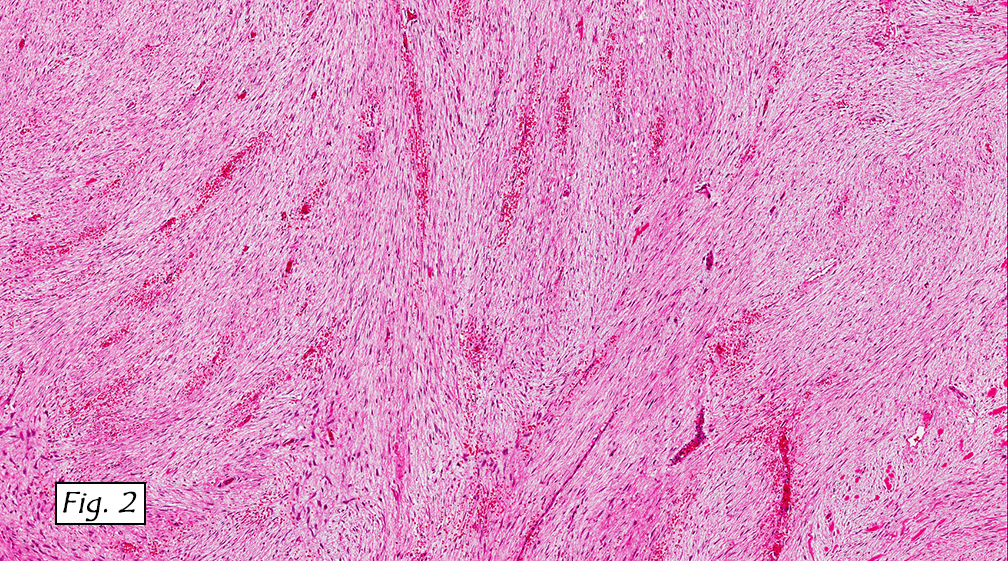
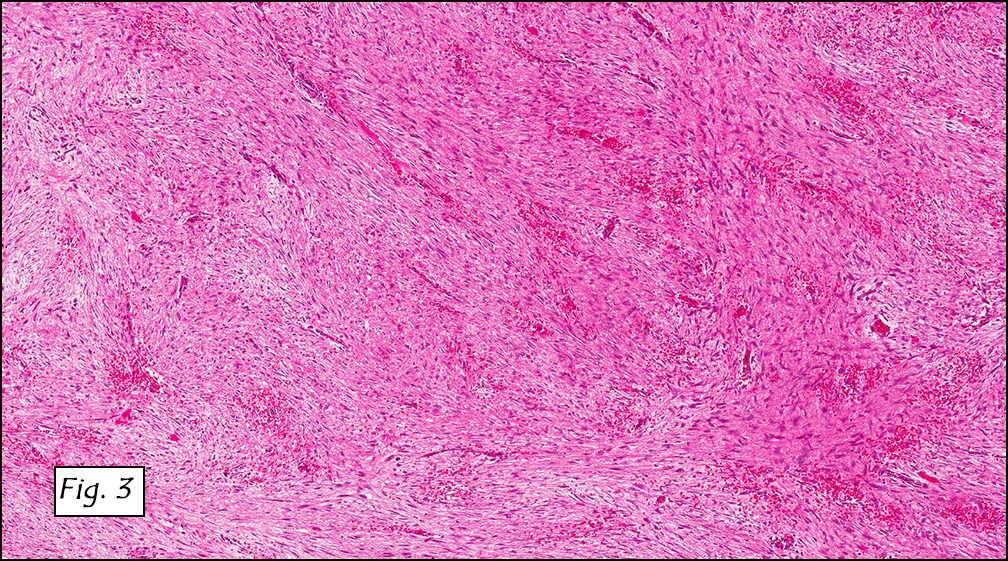
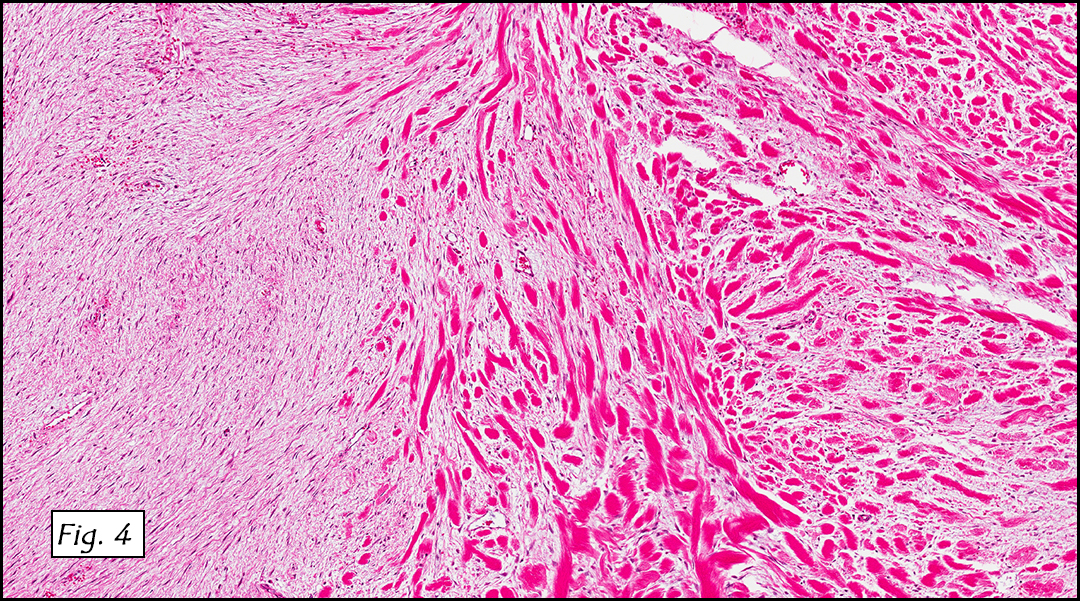
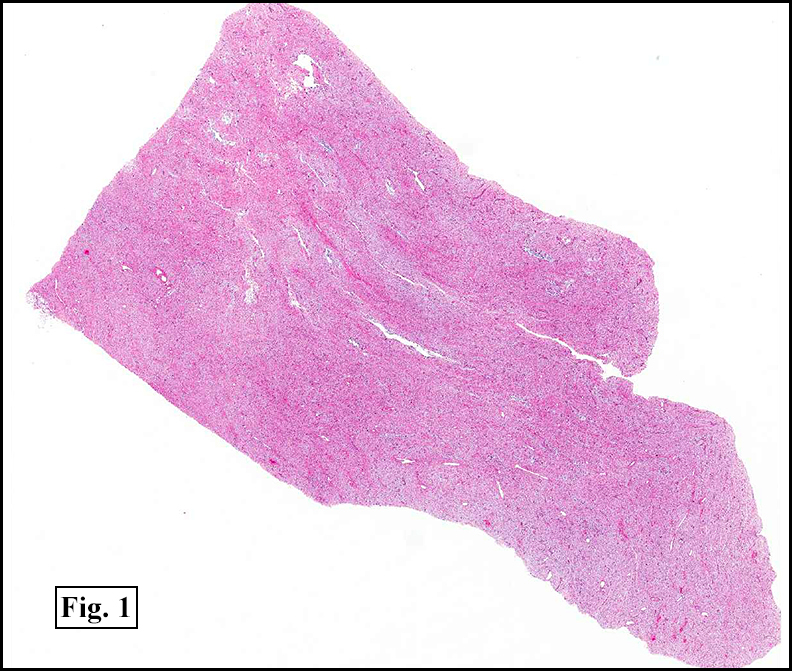
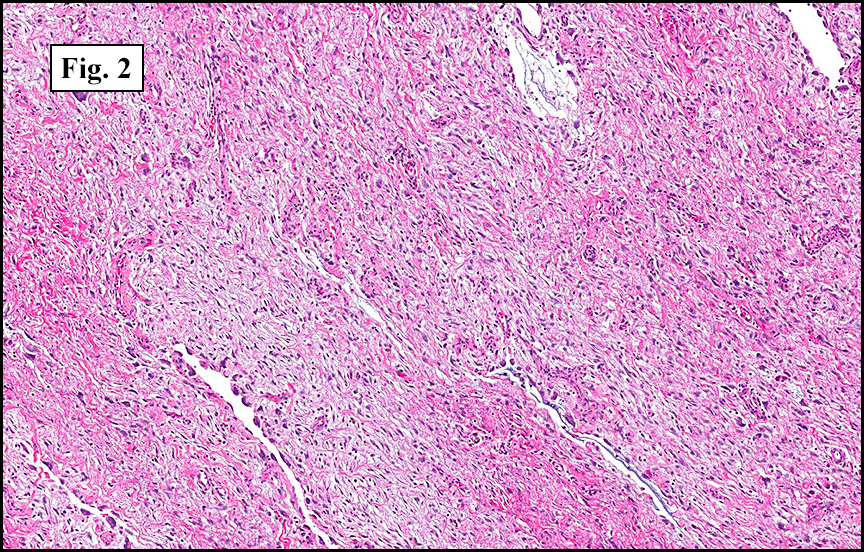
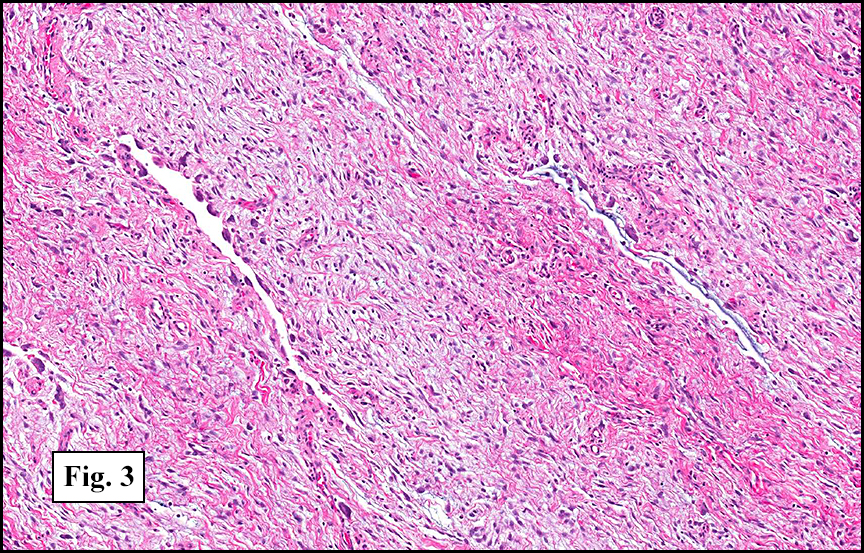
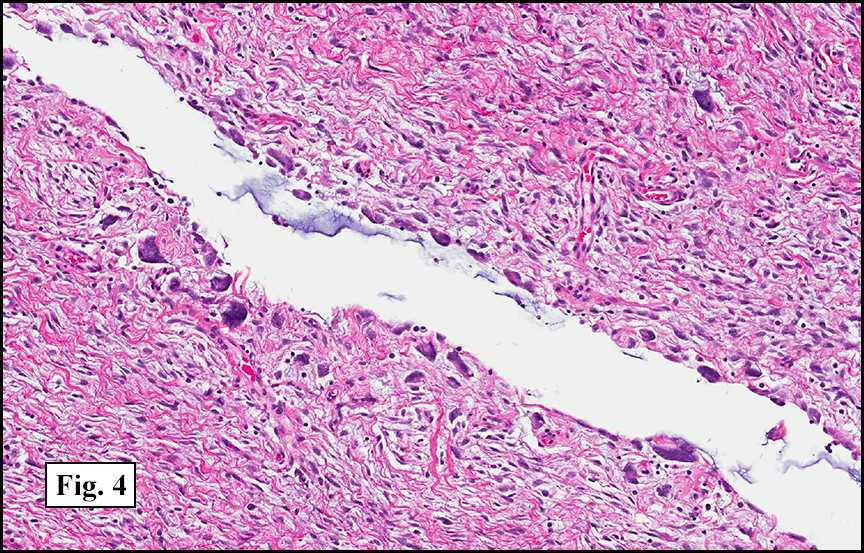
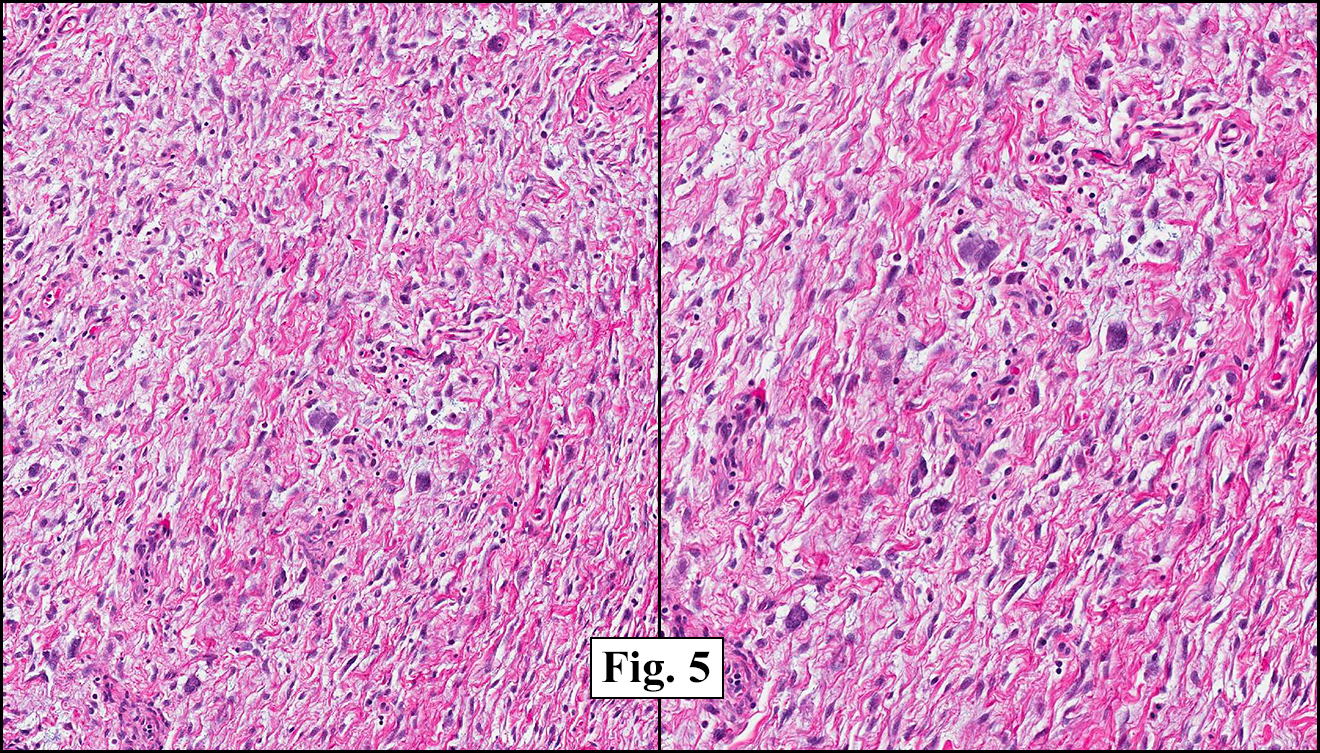
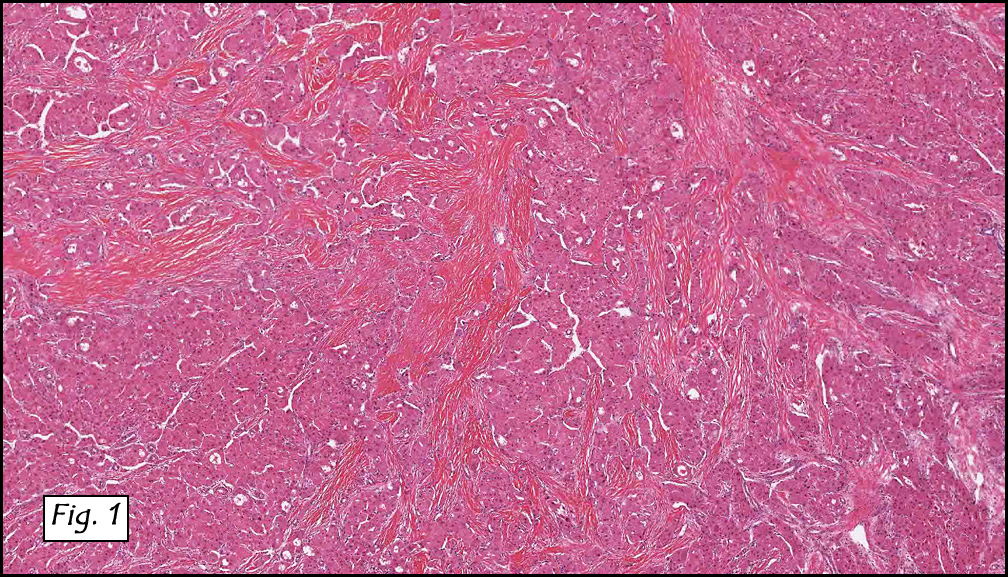
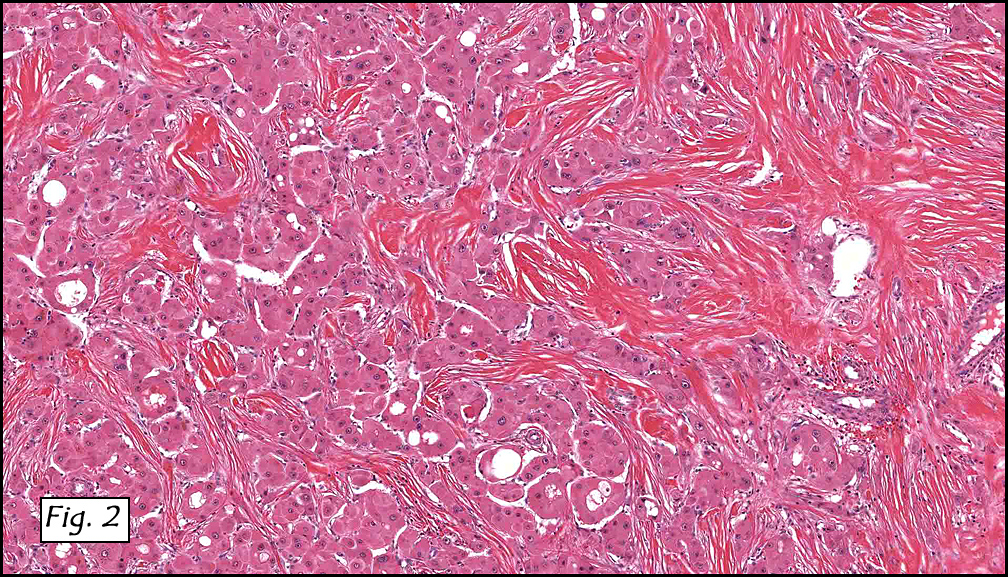
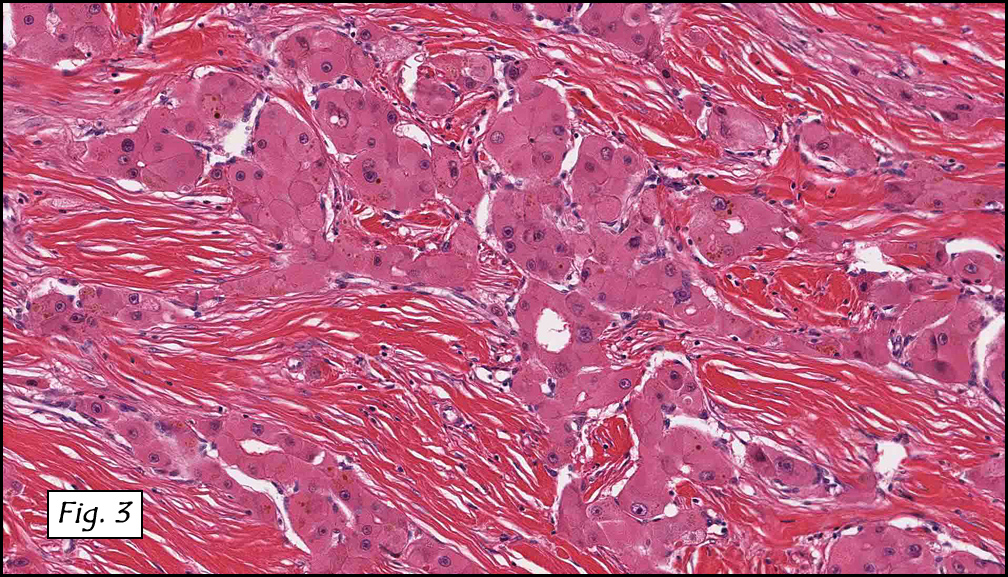
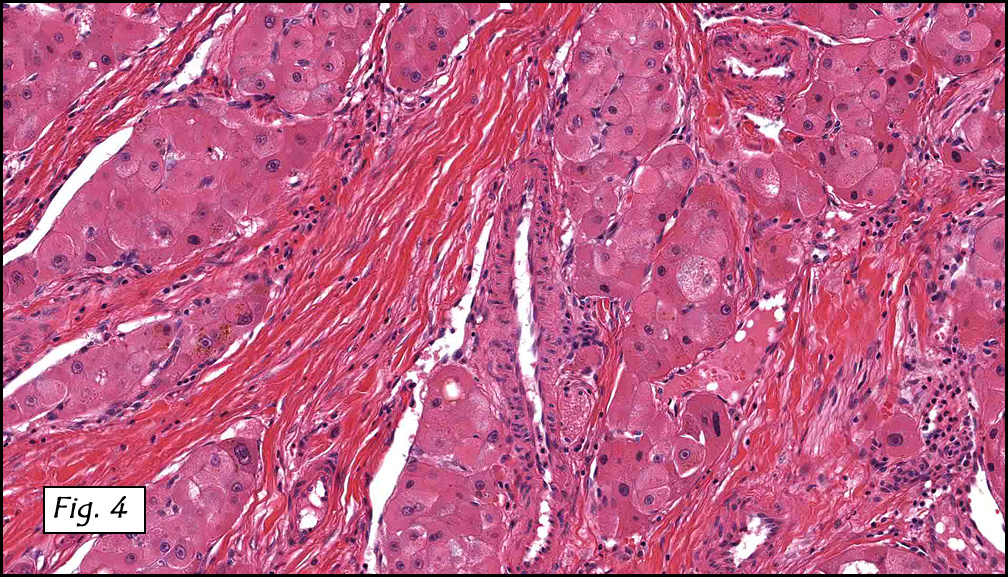
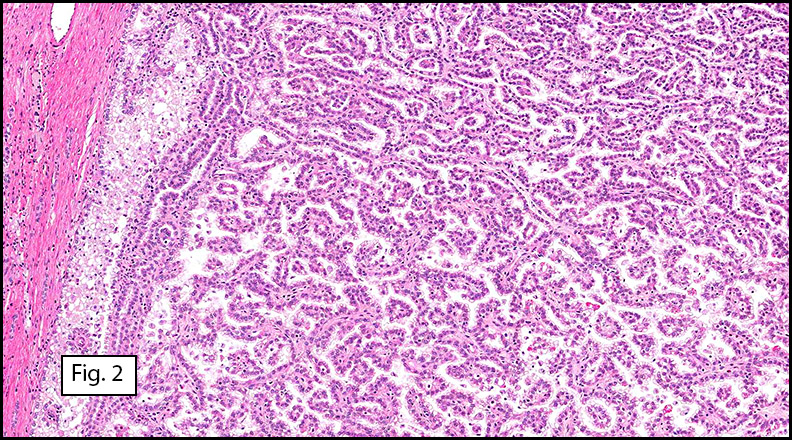
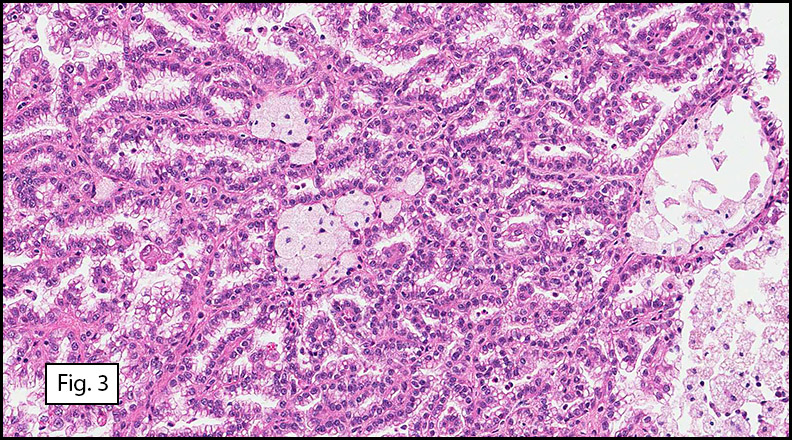
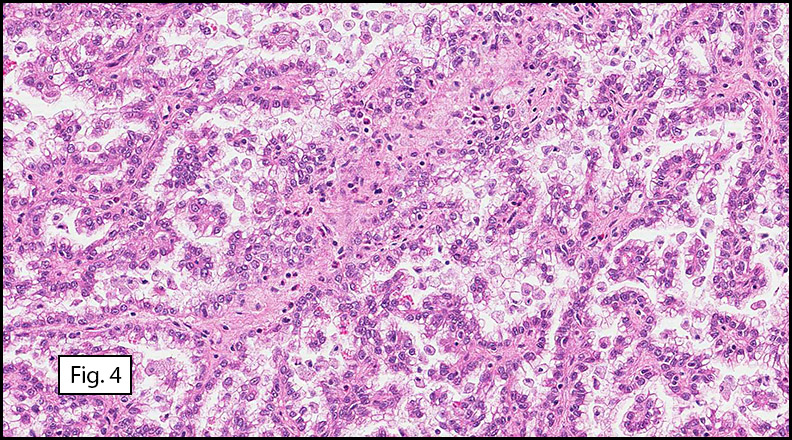
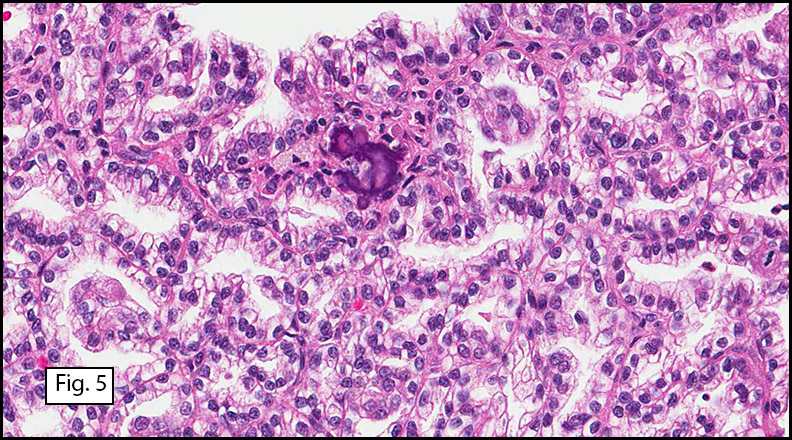
Grossly the tumor was 3 cm in diameter, well-circumscribed and encased within surrounding fat. The cut surface was gritty with focal cystic changes and peripheral fibrosis. Microscopically, the tumor had well defined margins with a thin fibrous capsule and focal calcifications (Fig. 1). The tumor consisted of spindled and polygonal cells arranged in alveolar and fascicular patterns (Fig. 2,3) with intervening dense, hyalinized collagen (Fig. 4). The cells displayed clear to eosinophilic cytoplasm with indistinct borders (Fig. 5) and round to oval nuclei demonstrating large, prominent nucleoli and coarse chromatin (Fig. 6). Mitotic figures were not identified and there was no evidence of necrosis. Immunohistochemistry revealed diffuse positivity for S100, HMB45, and vimentin with focal positivity for melanA and tyrosinase (Fig. 7).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Diagnosis: “Clear Cell Sarcoma (Malignant Melanoma of Soft Parts)â€
Drew G. Davis, MSIV, and Donald R. Chase, M.D.
Department of Pathology and Human Anatomy, Loma Linda University and Medical Center, Loma Linda, California
California Tumor Tissue Registry, Loma Linda, California
Discussion: Clear cell sarcoma was first described by Dr. Franz M. Enzinger in 1965. Subsequently, several histologic and ultrastructural similarities with melanoma were noted including melanin pigment, presence of melanosomes, and immunohistochemical staining for S-100 and HMB-45. These findings resulted in the moniker malignant melanoma of soft parts. Despite these similarities clear cell sarcoma and melanoma appear to be two different entities. Recent chromosomal analyses have demonstrated a characteristic translocation in clear cell sarcoma, t(12;22), resulting in a novel fusion gene product which is not found in melanoma.
A majority of cases are localized in the extremities, predominantly the lower legs, with the ankle being the most commonly affected site. Tumors have been noted on the trunk and neck, but intra-abdominal tumors are exceedingly rare. The tumors are intimately involved with adjacent tendon sheaths and aponeuroses and rarely involve the epidermis although occasional dermal extension has been noted.
Most clear cell sarcomas arise between the ages of twenty and forty but they have been noted in a wider range from seven to eighty three. There is a slight female predominance. Clinically they are recognized by gradually enlarging, occasionally painful masses present for months to years. Grossly, the rubbery, firm tumors are lobulated or multinodular and are often encapsulated or partially circumscribed. The cut surface is gray-white and homogeneous with occasional foci of necrosis or hemorrhage. Pigmentation may be observed in some cases.
Histologically, clear cell sarcoma is distinguished by alveolar and fascicular patterns of spindled to polygonal cells with clear to eosinophilic cytoplasm separated by characteristic collagenous septa. The nuclei display vesicular chromatin and one or more prominent nucleoli. Melanin pigment may be seen in up to 50% of cases. Occasional multinucleated giant cells are identified and rare mitoses are seen as well. Immunohistochemical stains are a useful adjunct in the diagnosis and show a similar staining pattern to that of melanoma with varying degrees of S-100, HMB-45, Melan-A and vimentin positivity. Metastatic tumor deposits typically show a similar histologic pattern but increased nuclear pleomorphism, disorderly growth and multiple foci of hemorrhage and necrosis have been observed.
The differential diagnosis includes sarcomas with predominantly fascicular growth patterns, including synovial sarcoma and fibrosarcoma, as well as melanin producing tumors such as cellular blue nevi and paraganglioma-like dermal melanocytic tumor (PDMT). The cytologic features of clear cell sarcoma including clear cytoplasm and prominent melanoma-like nucleoli, along with its immunophenotypic profile, differentiate it from other sarcomas. More important is the distinction between it and other melanocytic lesions. Although the survival rates of both melanoma and clear cell sarcoma are poor, the clinical management differs drastically. The location of the tumor, especially its investiture in adjacent tendons and aponeuroses, and its general lack of epidermal and dermal involvement are helpful clues. The uniform, spindled appearance of clear cell sarcoma also contrasts with the epidermoid appearance of nodular melanomas. Cytologic features are useful in distinguishing clear cell sarcoma from cellular blue nevi, which typically have smaller, pinpoint nucleoli and lack atypia. As a last resort, molecular genetic analysis for the t(12;22) translocation will establish the diagnosis of clear cell sarcoma. Paraganglioma-like dermal melanocytic tumors may also show cells with clear to eosinophilic cytoplasm but the presence of zellballen-like nests, low grade nuclei and their traditional location in the dermis set them apart from clear cell sarcomas.
The established treatment of choice is surgical excision with wide margins. Chemotherapy and radiation for both local and metastatic control have been used with little success. Metastases are common and affect up to 67% of patients, often years after the primary excision. Prognostic factors include necrosis, increased tumor size and the presence of metastases.
Suggested Reading:
Enzinger FM. Clear cell sarcoma of tendons and aponeuroses: An analysis of 21 cases. Cancer. 1965; 18:1163-1174.
Chung EB, Enzinger FM. Malignant melanoma of soft parts: A reassessment of clear cell sarcoma. Am J Surg Pathol. 1983; 7(5):405-13.
Lucas DR, Nascimento AG and Sim FH. Clear cell sarcoma of soft tissues: Mayo Clinic experience with 35 cases. Am J Surg Pathol. 1992; 16(12):1197-1204.
Montgomery EA, Meis JM, Ramos AG, Frisman DM and Martz KL. Clear cell sarcoma of tendons and aponeuroses: A clinicopathologic study of 58 cases with analysis of prognostic factors. Int J Surg Pathol. 1993; 1(2):89-100.
Langezaal SM, Graadt von Roggen JF, Cleton-Jansen AM, Baelde JJ and Hogendoorn PCW. Malignant melanoma is genetically distinct from clear cell sarcoma of tendons and aponeuroses (malignant melanoma of soft parts). Br J Cancer. 2001; 84(4):535-8.
Weiss S, Goldblum J. Enzinger and Weiss’s Soft Tissue Tumors (5th edition). Philadelphia: Mosby/Elsevier Inc. 926-34, 2008.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}