History: A 70-year-old retired sandblaster underwent a left lower lobectomy for lung cancer. He had stopped smoking one year earlier, after smoking 1.5 packs per day for approximately 50 years. Four years later, two new nodules were discovered, resulting in a right lower lobectomy.
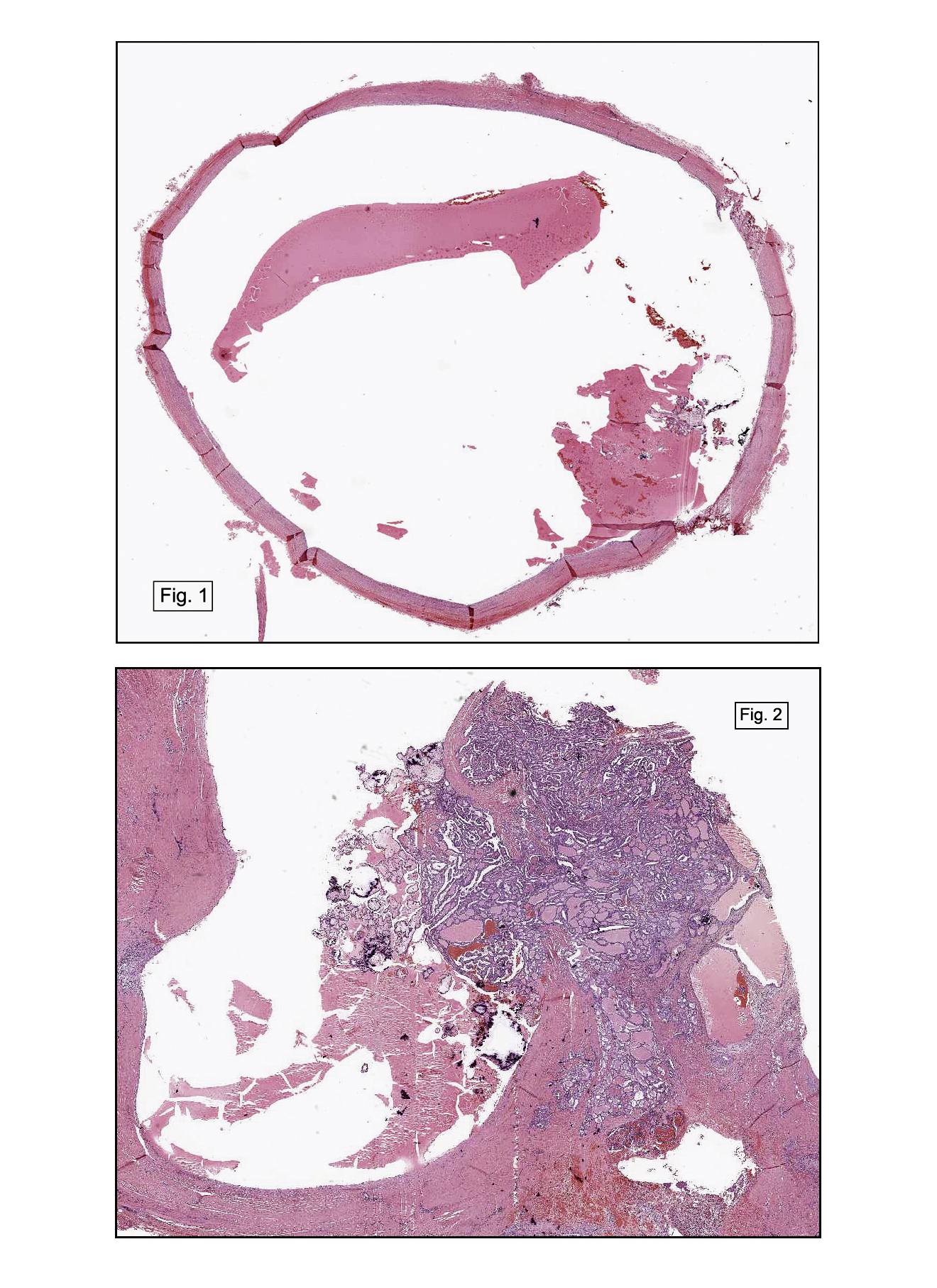
Gross examination revealed a dominant, ill-defined, marbled, gray-and-white tan 6 cm tumor which involved the pleural surface and showed central cystic degeneration, as well as numerous smaller satellite lesions of similar appearance. Three hilar lymph nodes were also identified.
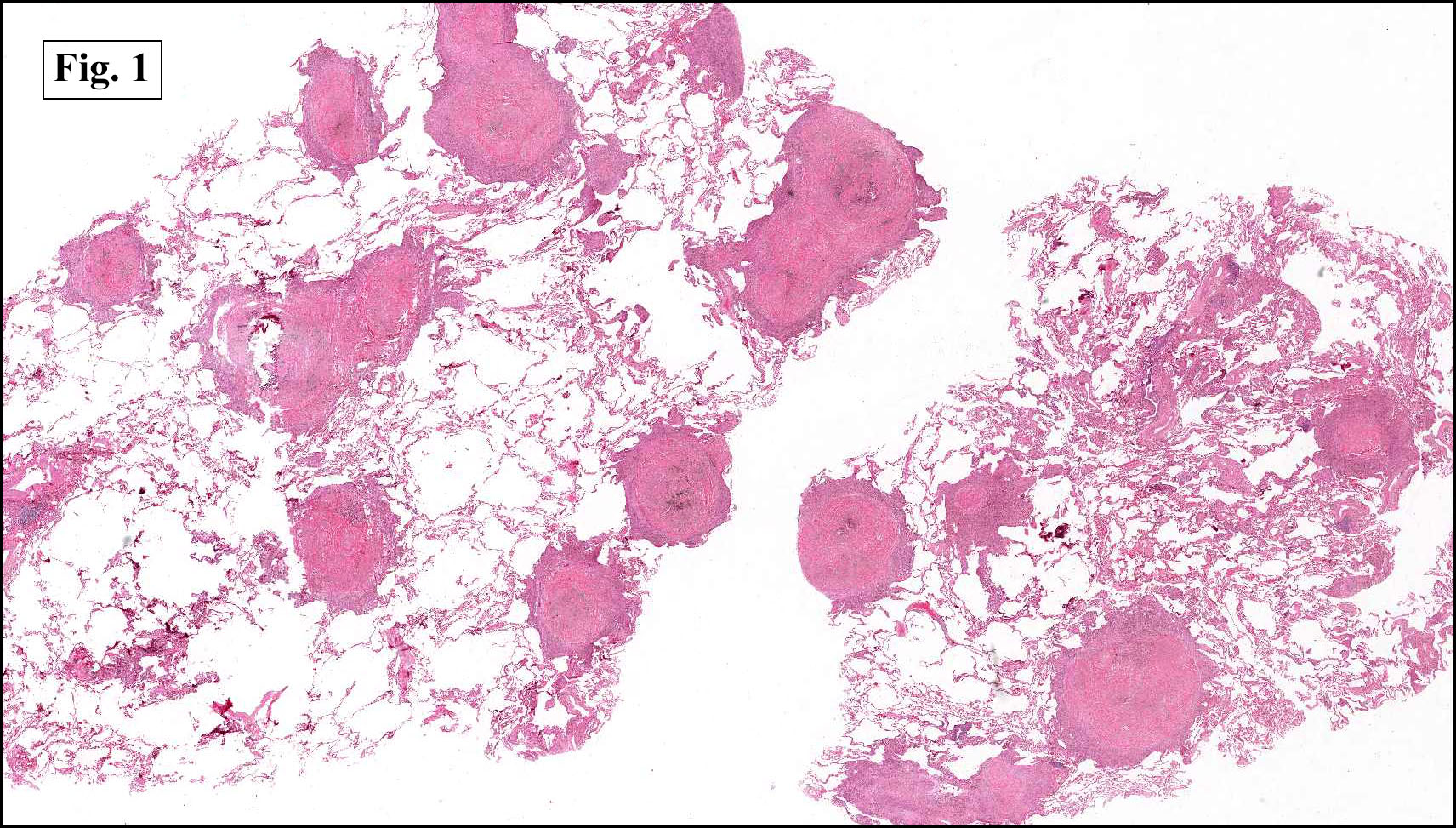
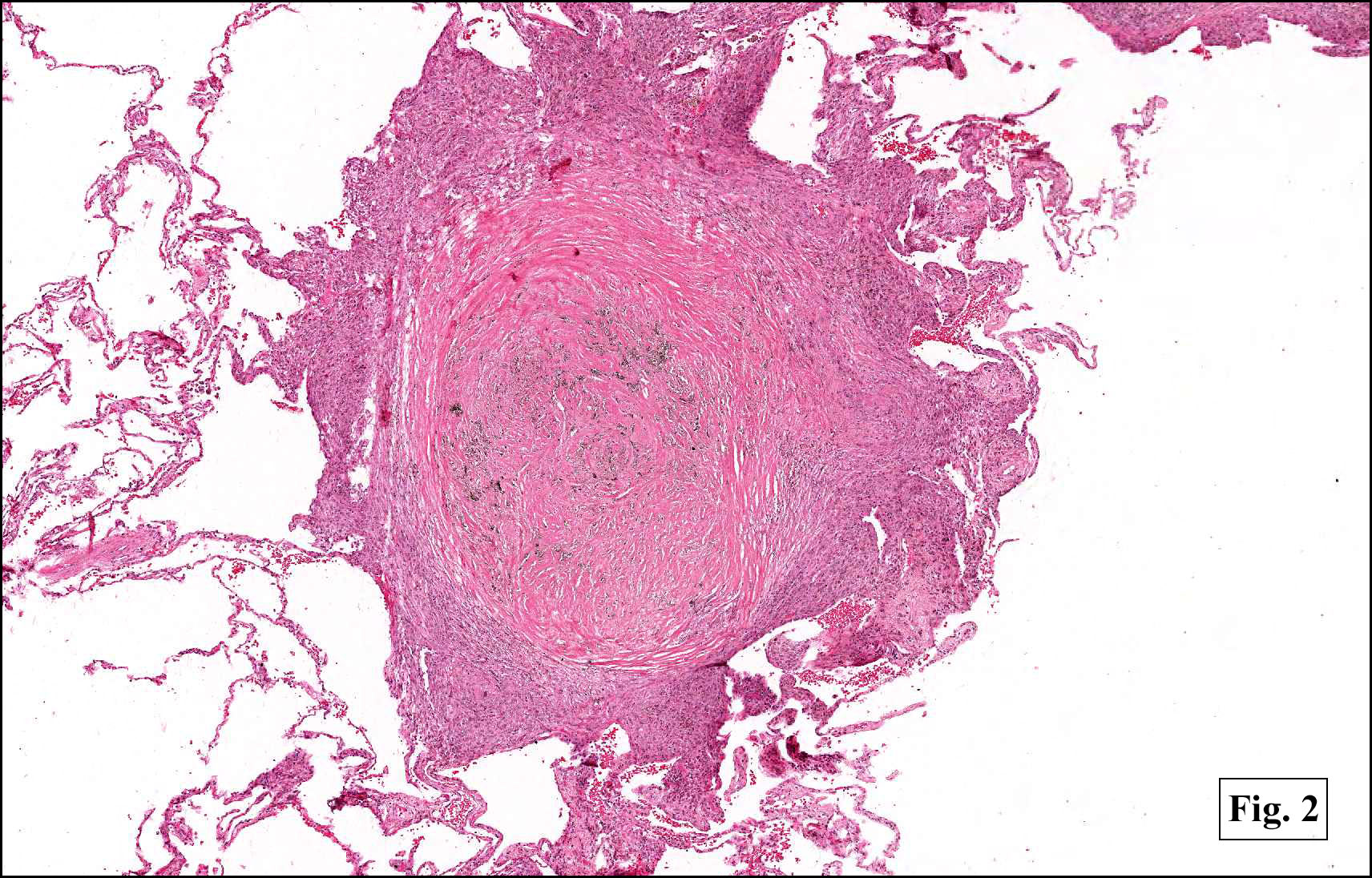
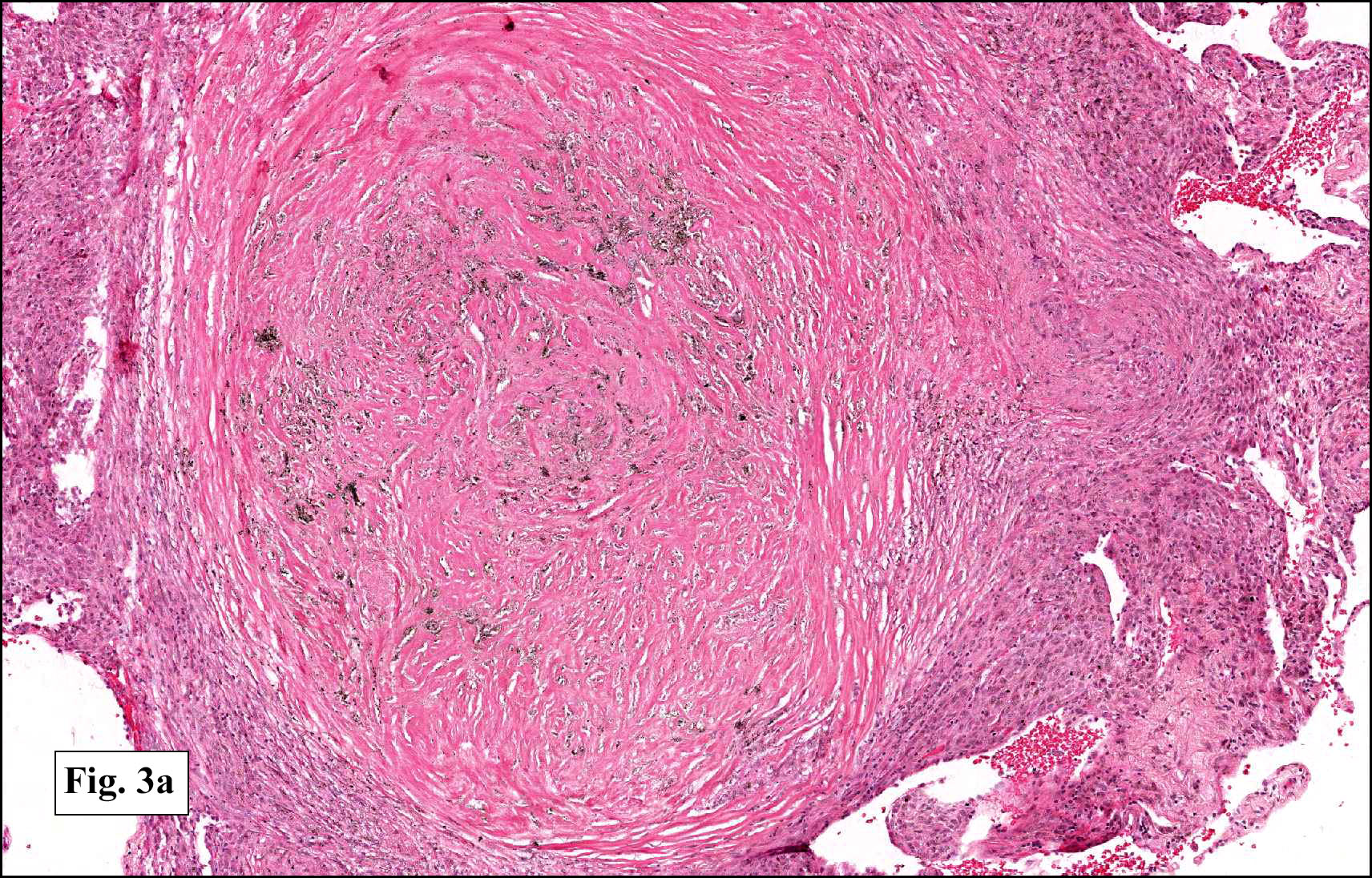
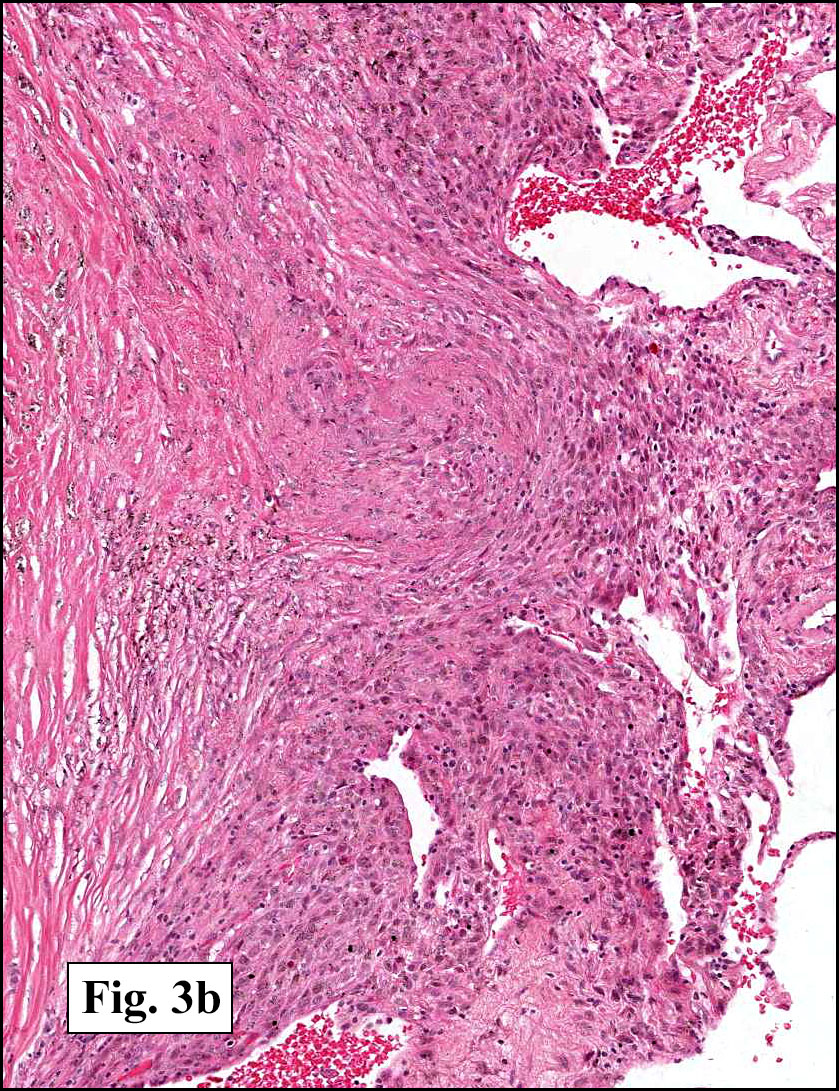
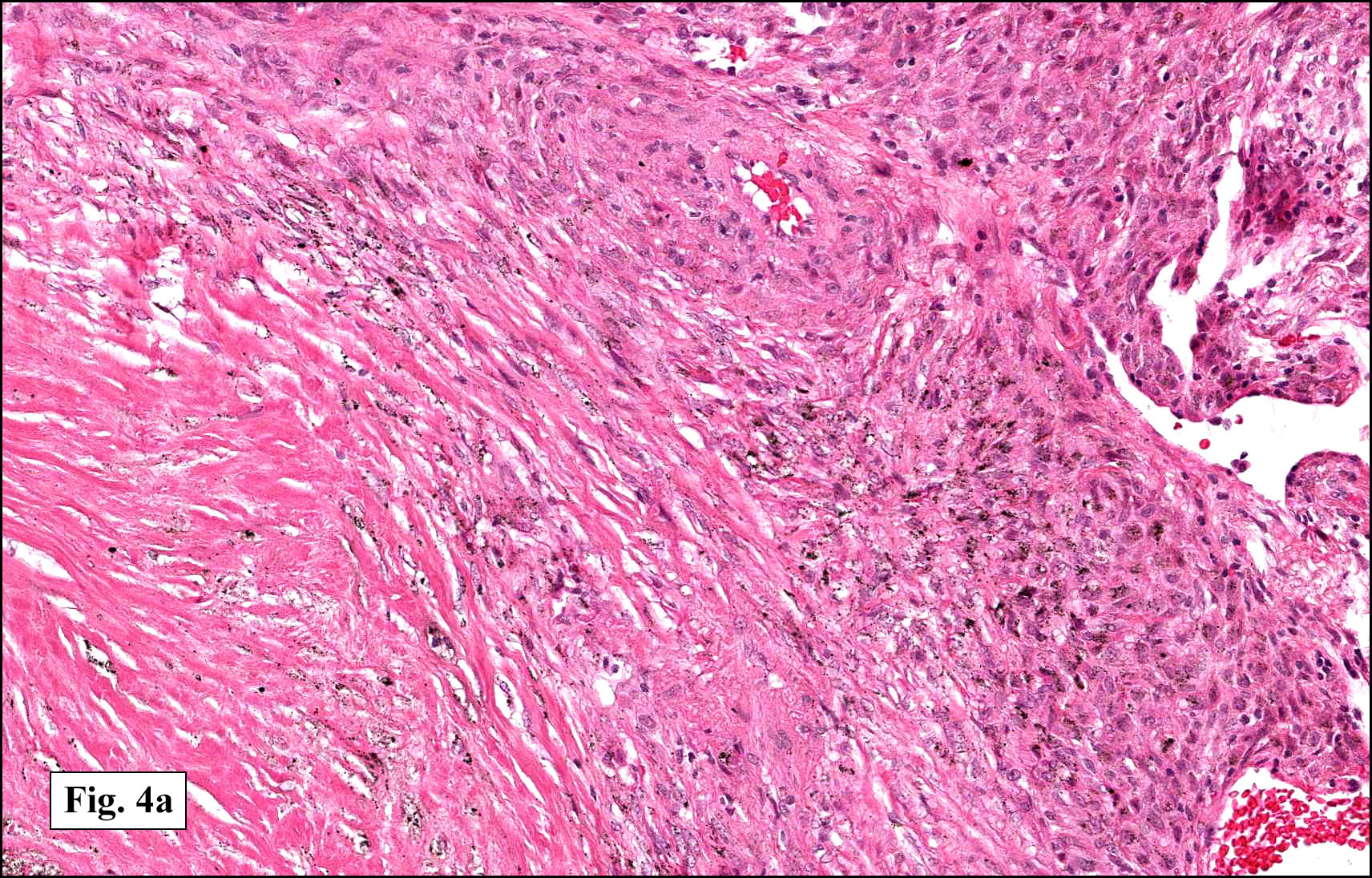
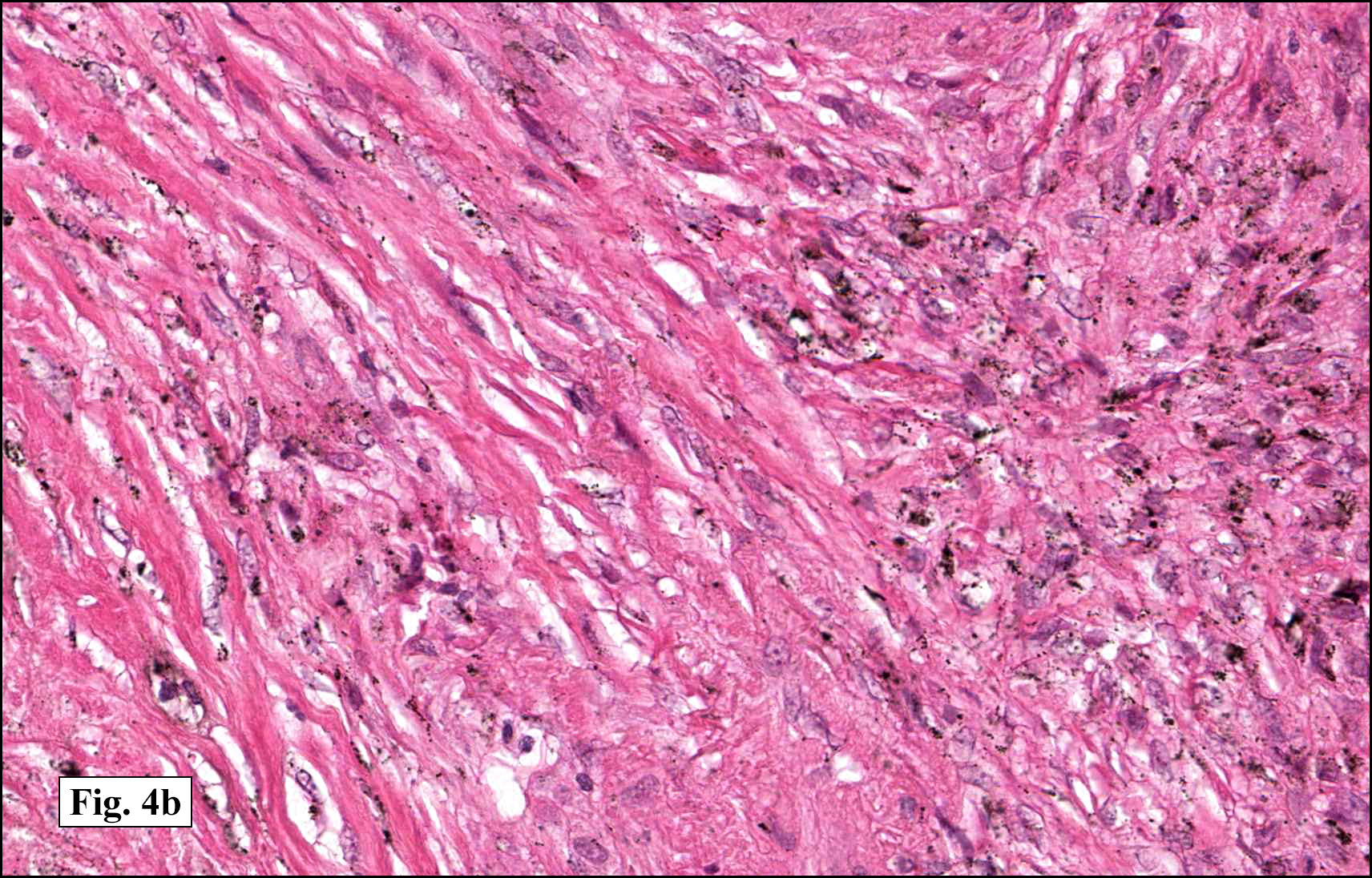
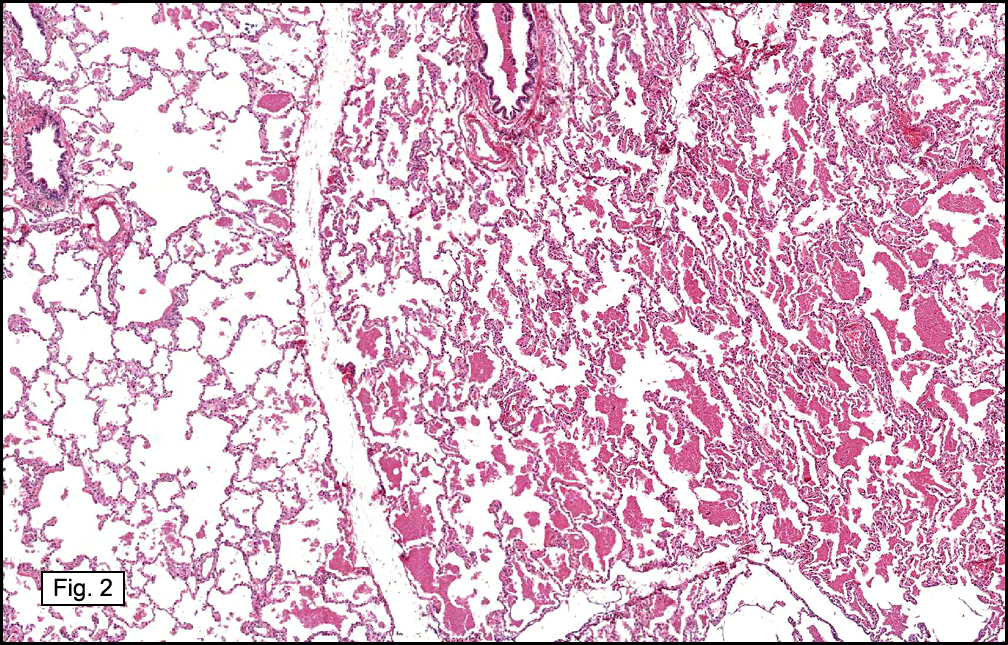
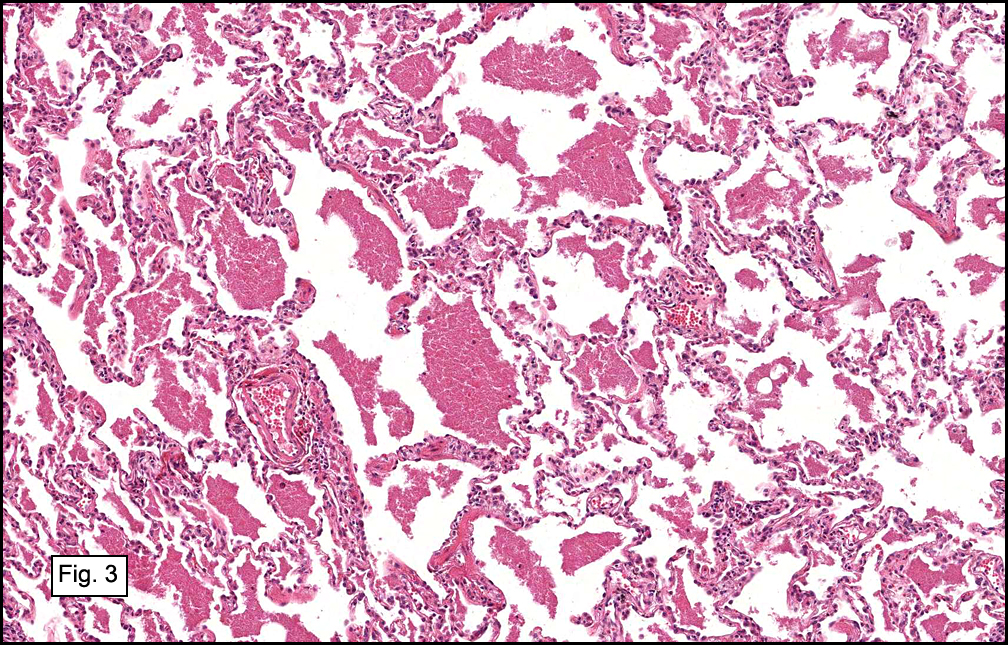
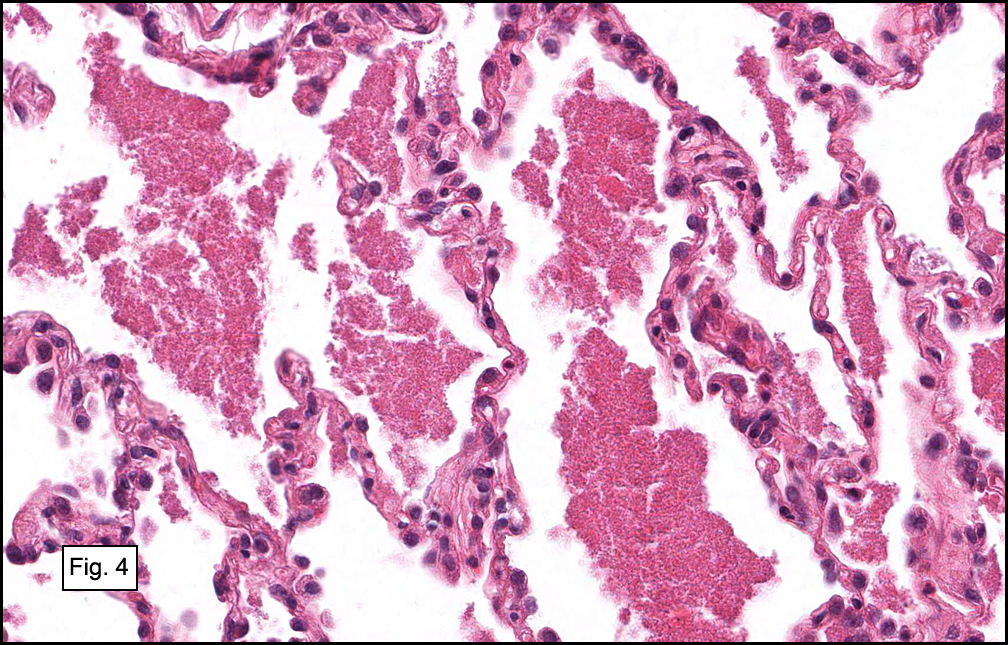
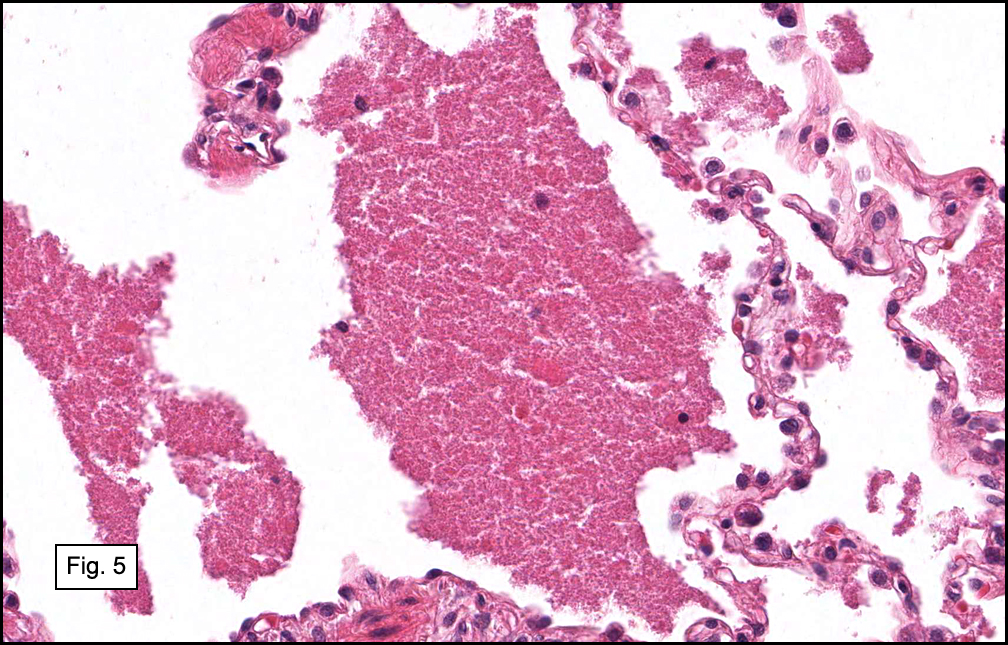
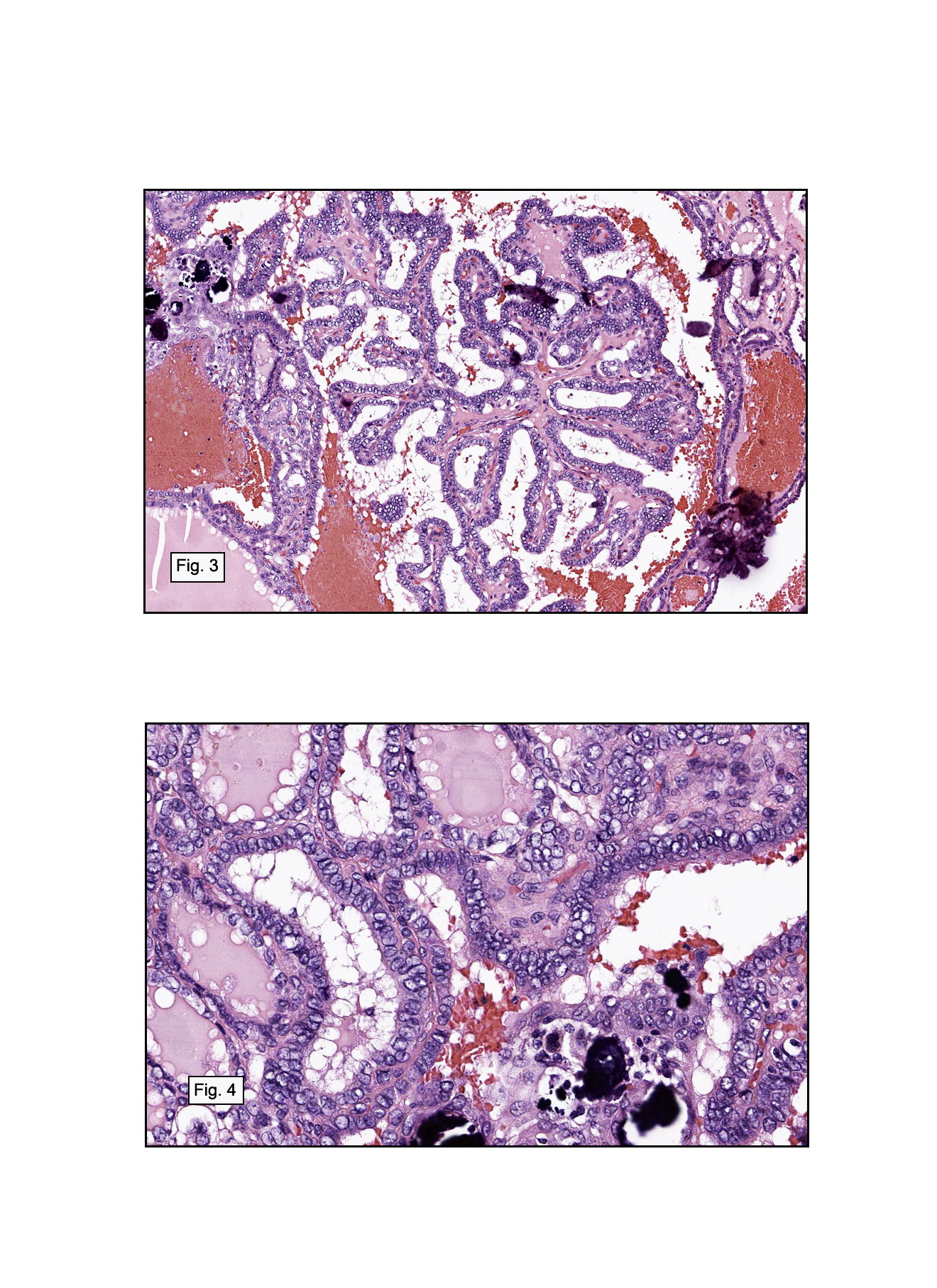
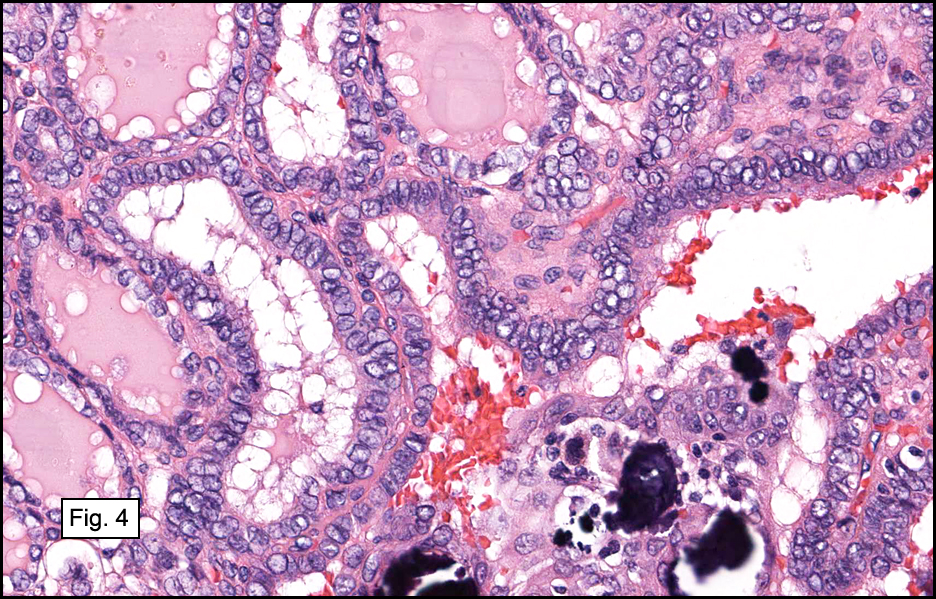
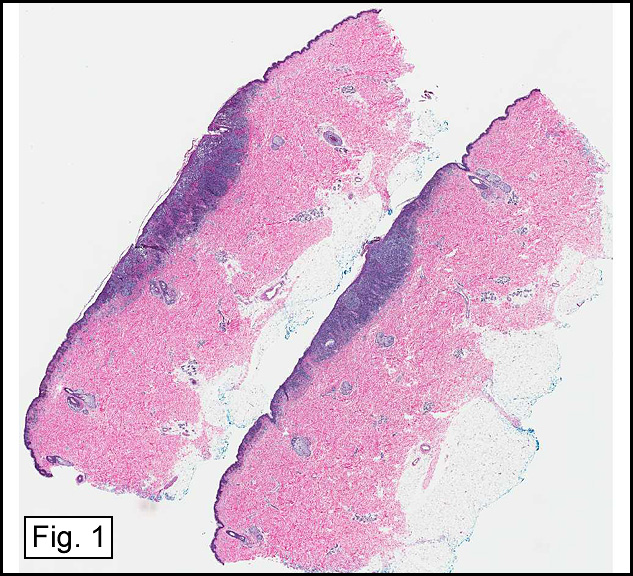
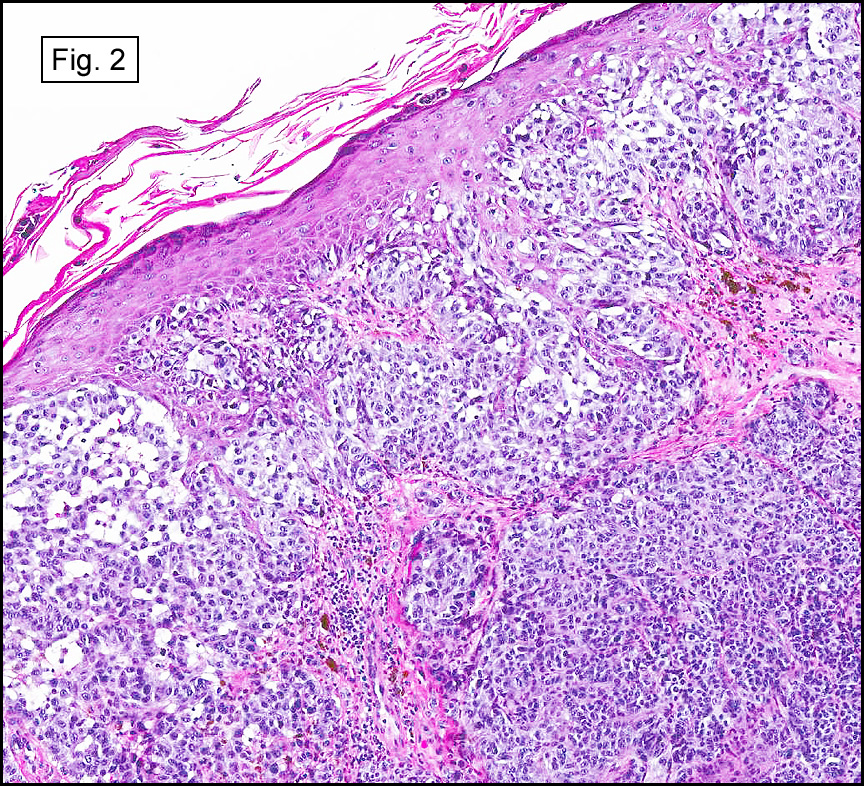
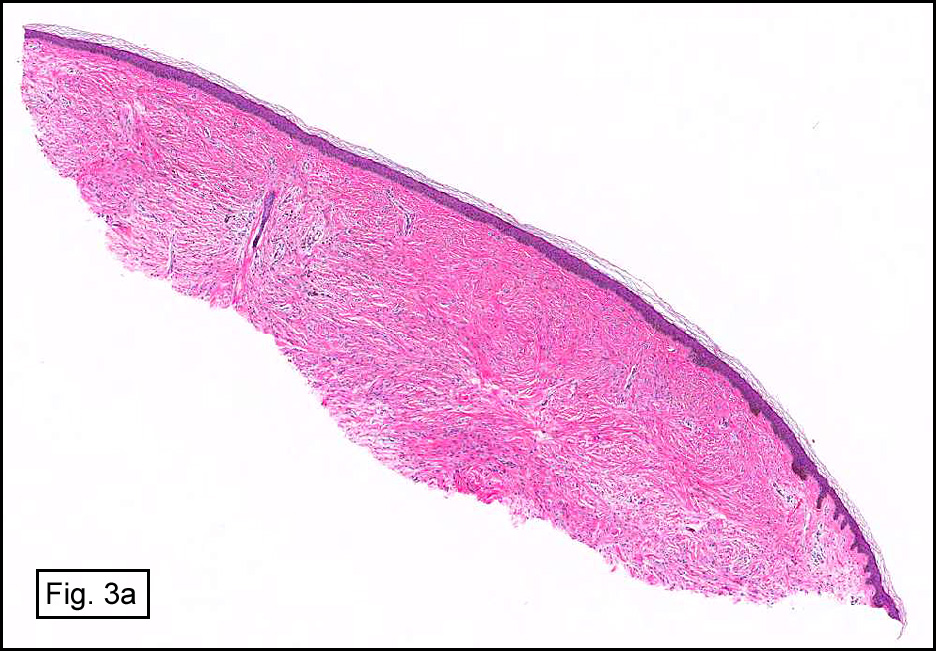
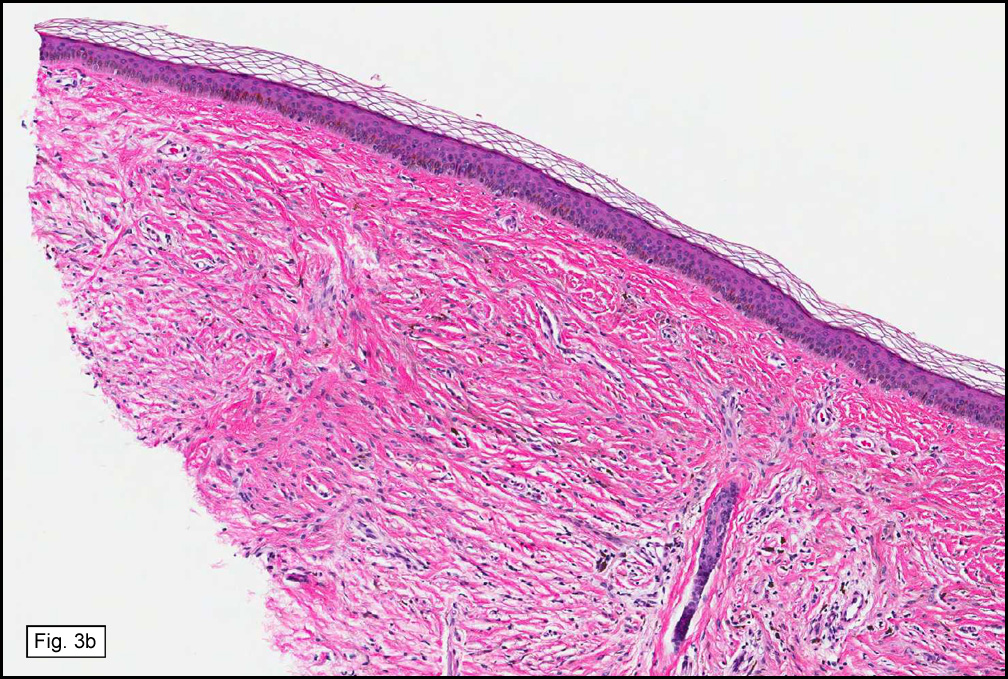
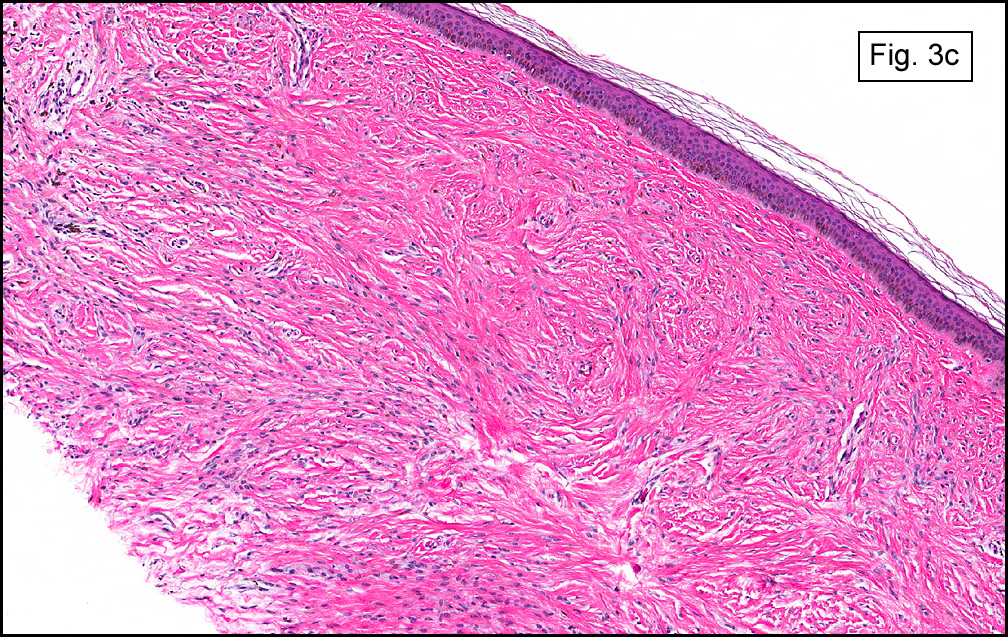
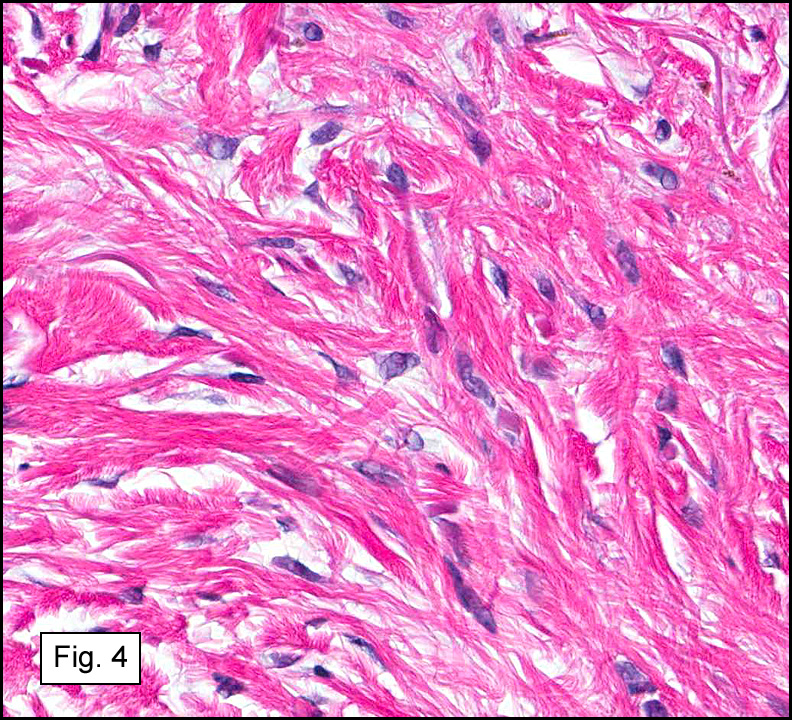
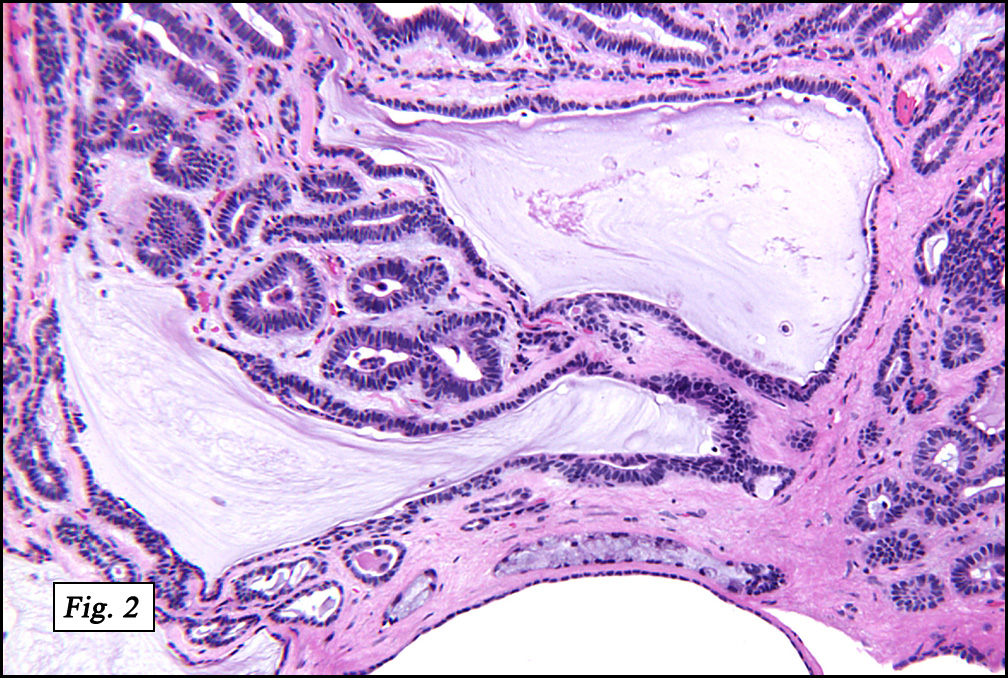
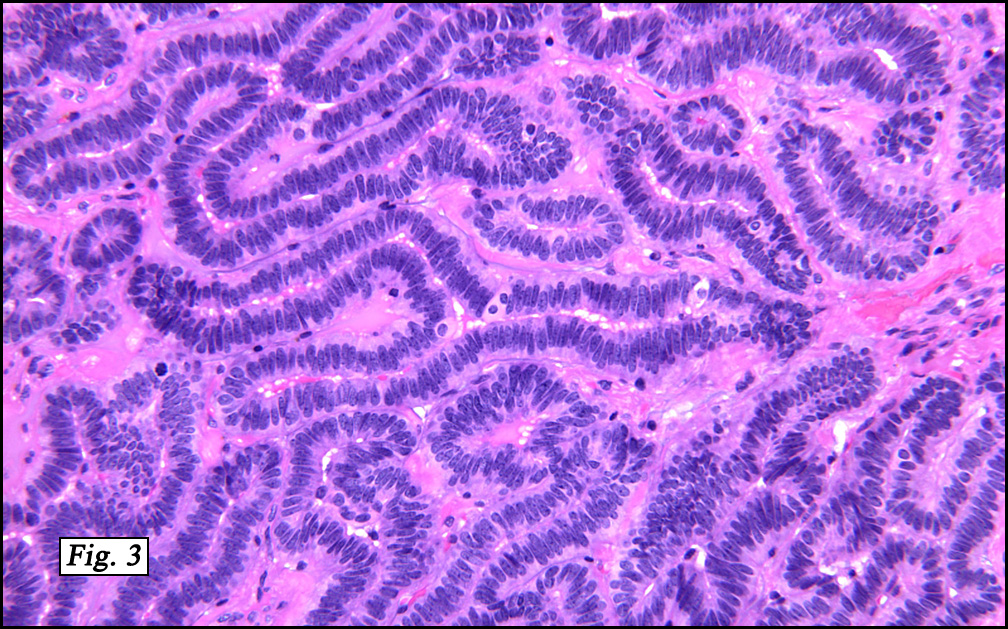
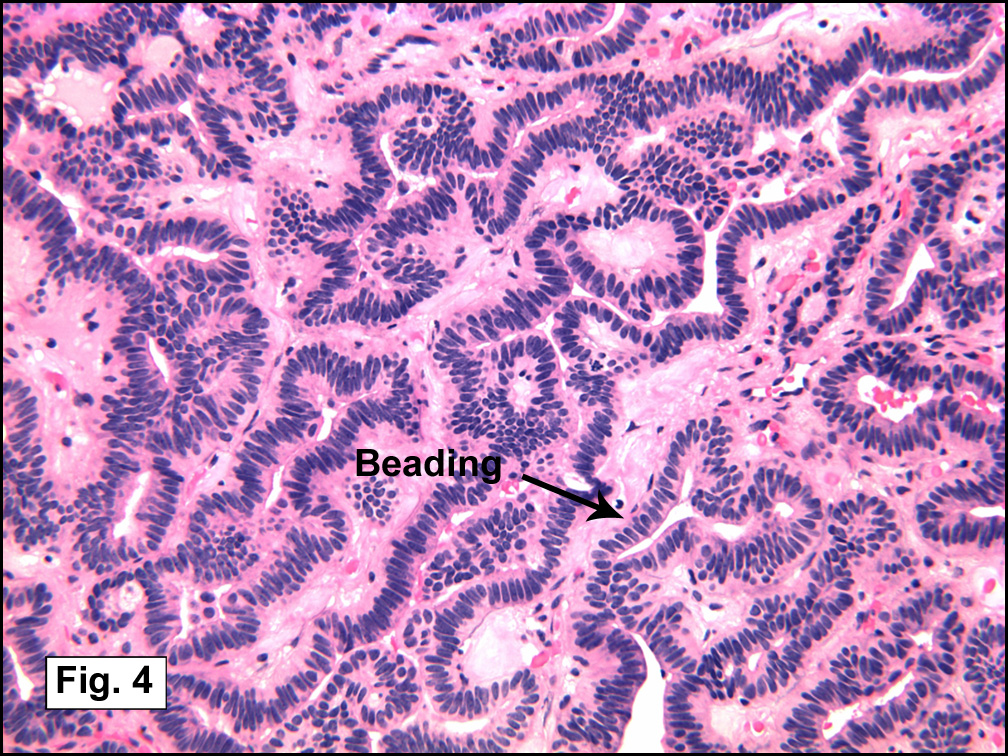
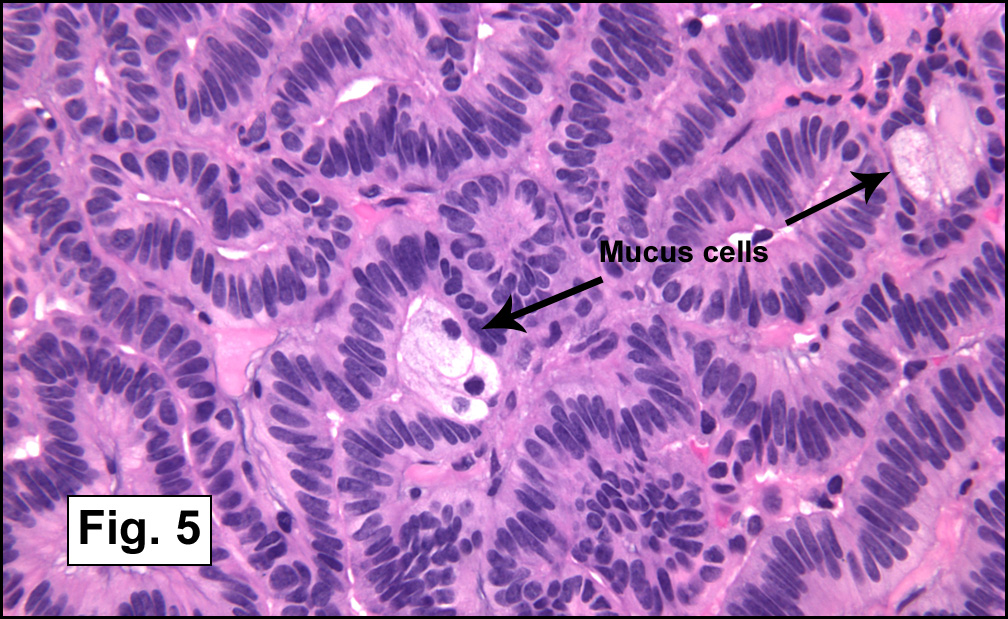
In some sections (not illustrated), squamous cell carcinoma was identified. The point of interest for us, however, was the background lung. Scattered throughout the parenchyma were numerous dense, homogenous nodules composed of brightly-eosinophilic, concentrically-layered material (Fig. 1, 2, 3a, 3b). Dust-filled macrophages were observed, as was emphysematous change (Fig. 4a, 4b). The eosinophilic nodules also involved the accompanying lymph nodes.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Diagnosis: “Silicosis Co-Existent With Pulmonary Adenocarcinomaâ€
Christina M. Birsan M.D., and Donald R. Chase, M.D.
Department of Pathology and Human Anatomy, Loma Linda University and
Medical Center, Loma Linda, California
California Tumor Tissue Registry, Loma Linda, California
Discussion: Silicosis, a chronic lung disorder, is characterized by progressive development of parenchymal nodules and pulmonary fibrosis as a result of inhalation of crystalline silica. Most cases are related to exposure to quartz, the most abundant mineral in the earth’s crust and also the most abundant form of crystalline silica. Identified in Egyptian mummies, silicosis is the oldest recognized occupational lung disease, with exposure occurring in various rock-cutting, sand-blasting, and ceramics industries.
Clinically, silica-induced lung disease may be acute, accelerated, or chronic. Acute silicoproteinosis is similar to pulmonary alveolar proteinosis, and occurs in cases of heavy exposure. Patients are generally symptomatic within 3 years of exposure, and most cases are fatal. When the disease manifests between 3 to 10 years after exposure, accelerated silicosis is the suggested term. Chronic (classic or nodular) silicosis is the most common form, usually occurring decades after exposure to relatively low levels of silica. Classically, it requires at least 20 years after onset of exposure. It may manifest as simple silicosis (nodules 10 mm or less) or complicated silicosis (nodules greater than 1 cm). Simple silicosis is generally asymptomatic until advanced stages when dyspnea develops. Complicated silicosis results from coalescing smaller nodules over time (also called conglomerate silicosis). Cough and sputum production may occur, but are often the result of chronic bronchitis or concurrent infections. Crackles and digital clubbing are rare in silicosis, and suggest alternate diagnoses. Infections may complicate the process (mycobacterial infections in particular) and can cause fever and weight loss. Severe, longstanding silicosis and fibrosis may cause signs of cor pulmonale. Rheumatoid factor, antinuclear antibodies, serum immune complexes, and polyclonal increase in immunoglobulins may be detected in some cases. In fact, connective tissue disorders (most commonly scleroderma, rheumatoid arthritis, and systemic lupus erythematosus) have been associated with silica exposure. Signs and symptoms in those cases are more specific to the individual entities, and pulmonary silicosis is not always radiologically detectable. Significant associations between silicosis and lung cancer have been reported as well.
Radiologically, classic silicosis manifests as multiple nodules which mostly involve upper lobes. Best seen on CT scans, these nodules are well-circumscribed, uniform in size, and usually less than 5 mm in diameter. They may progress to form conglomerate masses, resulting in larger size. Complicated silicosis is usually bilateral and causes marked loss of lung volumes in the upper lobes, expansion and hyperinflation of the lower lobes, and superior retraction of the hila. Hilar lymph nodes are usually involved and may show a peripheral “eggshell†type calcification. Nodules have also been described in liver, spleen, bone marrow, and abdominal lymph nodes.
Grossly, chronic silicosis demonstrates firm, rounded, well-demarcated nodules, usually about 3 to 6 mm and slate gray to black. A cuff of pigmentation may be seen surrounding nodules on the pleural surface.
Microscopically, the same discrete nodules are apparent. Their formation begins with the accumulation of dust-filled macrophages along lymphatic routes, bronchovascular bundles, and in the pleura and septa. Early stages of disease may demonstrate only these accumulated macrophages. Increased reticulin and fibrosis in the sheets of macrophages represent developing nodules, which then enlarge and become discrete and round, replaced by dense lamellar collagen. They may become calcified, hyalinized, or may undergo central degeneration including cavitation and necrosis, in which case infection (especially tuberculosis) should be carefully ruled out. Polarized light reveals weakly birefringent silica and strongly birefringent round or oval silicate particles within nodules and in surrounding macrophages. If exposure stops, nodules may become fibrous with a few surrounding histiocytes. Emphysematous change is common in surrounding lung. Acute silicosis (silicoproteinosis) resembles pulmonary alveolar proteinosis (PAP), with an eosinophilic, granular, PAS-positive material filling the alveolar spaces. Unlike PAP, however, interstitial inflammation and fibrosis and irregular hyaline scars may be present. Silicotic nodules are usually poorly-formed or absent in such cases.
The pathogenesis of silicosis involves the inhalation of particles less than 10 micrometers in diameter, with the most pathogenic particles being approximately 1 micrometer. The intensity and duration of exposure are the most important factors, as the damage occurs once the silica particles are deposited in the lung. Direct cytotoxicity or the production of oxidants and other mediators may be involved, however the exact sequence of events is unknown. It appears that alveolar macrophages ingest the silica particles and die, perhaps liberating fibroblast-stimulating factors which promote fibrosis. Evidence also suggests that silica interferes with the ability of macrophages to inhibit the growth of mycobacteria, hence the association with tuberculosis. Toxic effects of silica on alveolar type 2 cells has been implicated in silicoproteinosis (acute silicosis).
The diagnosis of silicosis rests upon an appropriate exposure history, consistent radiologic findings, and the absence of other diseases which could explain the radiologic evidence. Observing silica, silicates, silicotic nodules, or increased dust in lymph nodes alone is not sufficient for the diagnosis, as virtually all adults have a small amount of silica in their lungs.
Unfortunately, no effective therapy exists once the inhalation of silica dust has occurred. Most affected patients are asymptomatic, however, and have a normal lifespan, with only a small percentage experiencing progressive respiratory disability.
Suggested Reading:
Travis W, Colby T, Koss M, et al. Non-neoplastic disorders of the lower respiratory tract. In King D, ed. Atlas of Nontumor Pathology. Washington, DC: American Registry of Pathology, Armed Forces Institute of Pathology; 2002.
Katzenstein AL III. Katzenstien’s and Askin’s Surgical Pathology of Non-neoplastic Lung Disease. 3rd edition. Philadelphia, PA: WB Saunders Company; 1997.
Hammar S. Pleural diseases. In: Dail D, Hammar S, eds. Pulmonary Pathology. 2nd ed. New York, NY: Springer-Verlag; 1994.
Erren TC, Morfeld P, Glende CB, et al. Meta-analyses of published epidemiological studies, 1979-2006, point to open causal questions in silica-silicosis-lung cancer researchâ€. Med Lav 2011 Jul-Aug; 102(4):321-35.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}