History: A 33 year-old woman presented with a palpable, 6 cm mass in the thigh. At surgery, it was found to deeply reside within the anterior compartment where it involved muscle. Osseous involvement was not seen. The excised specimen was poorly circumscribed, and had a cut surface that was fleshy tan-grey.
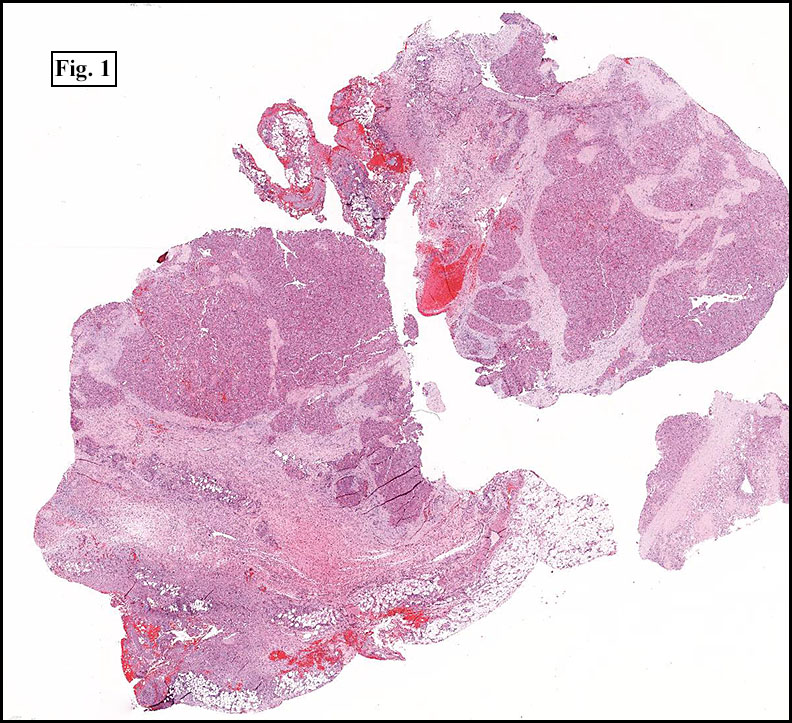
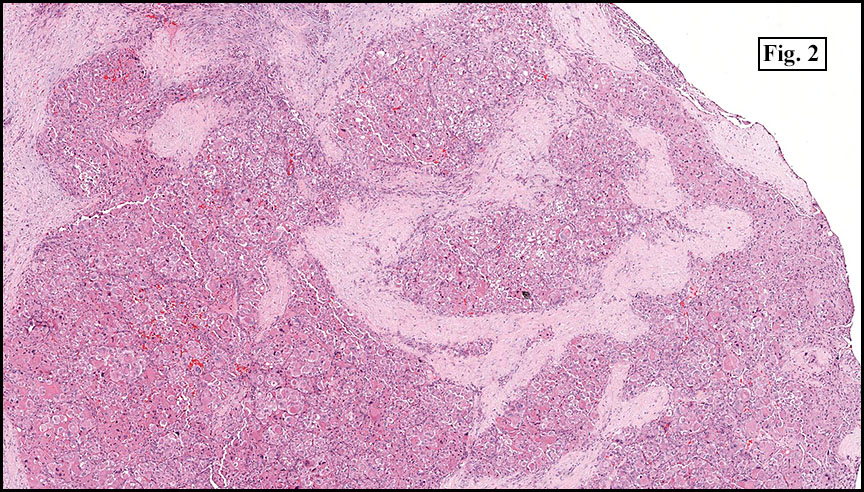
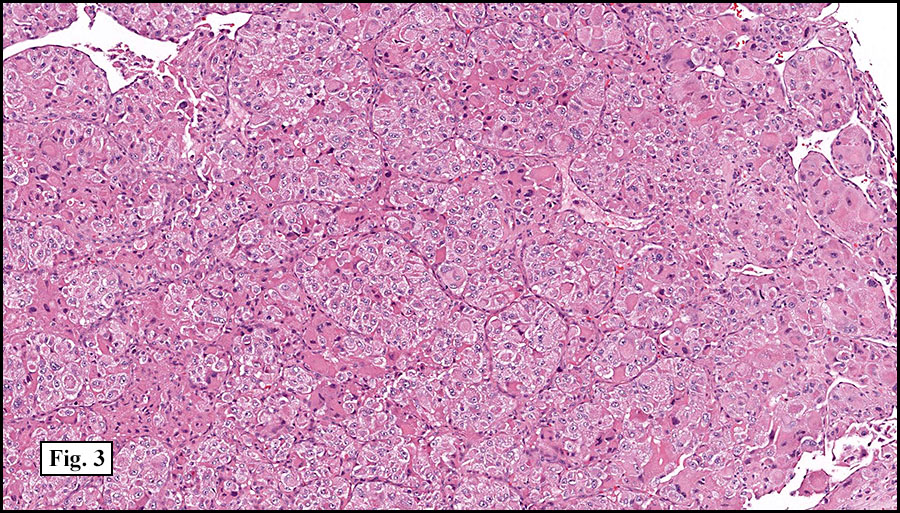
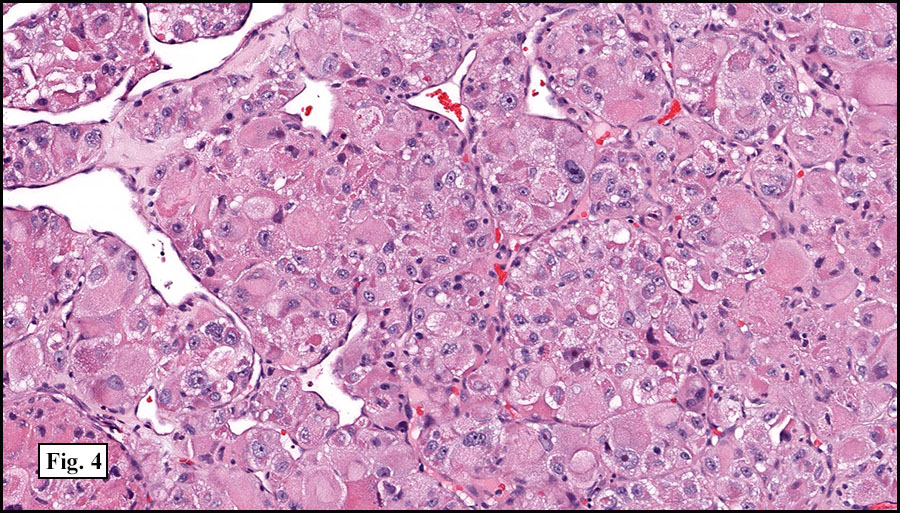
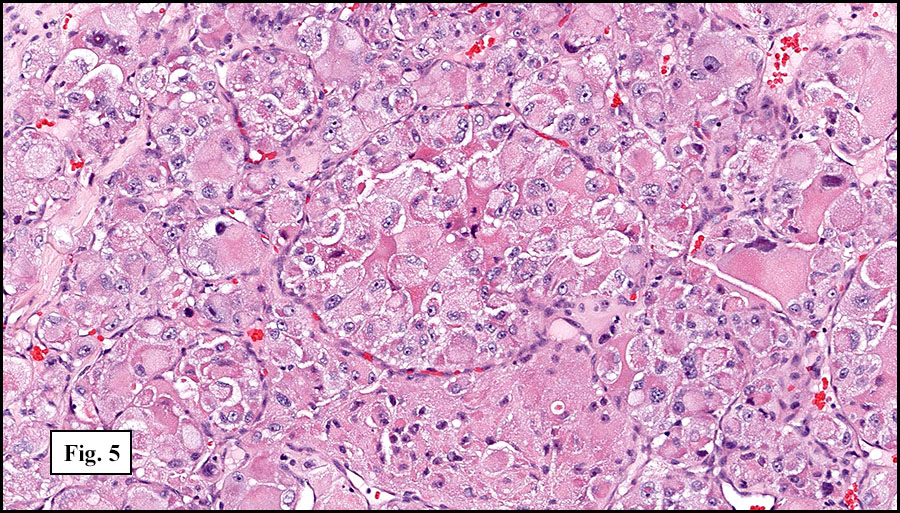
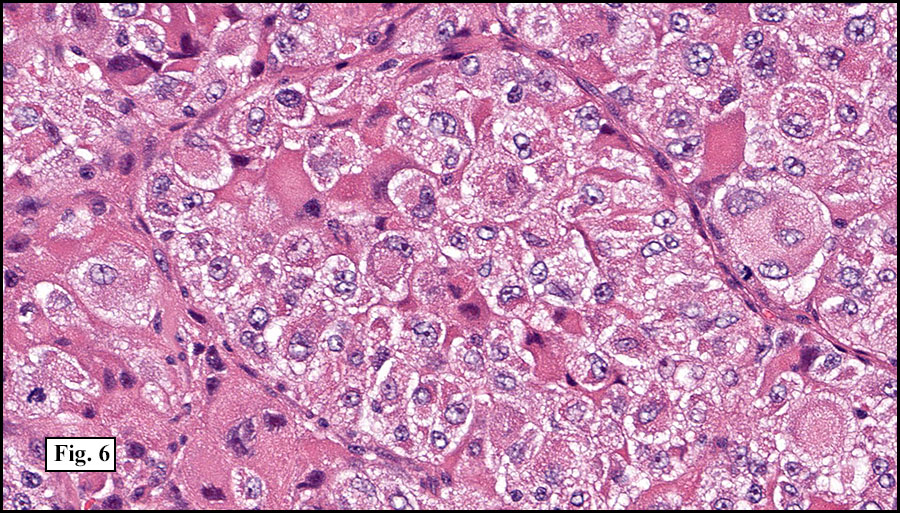
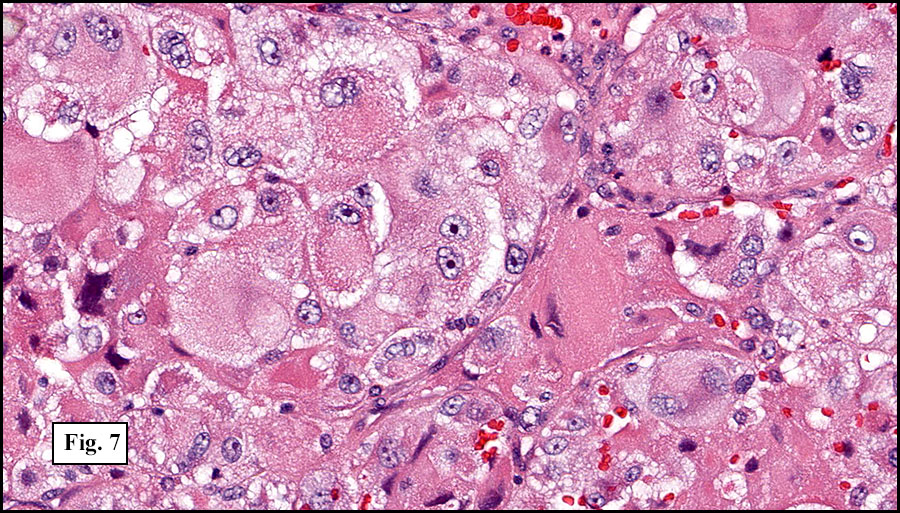
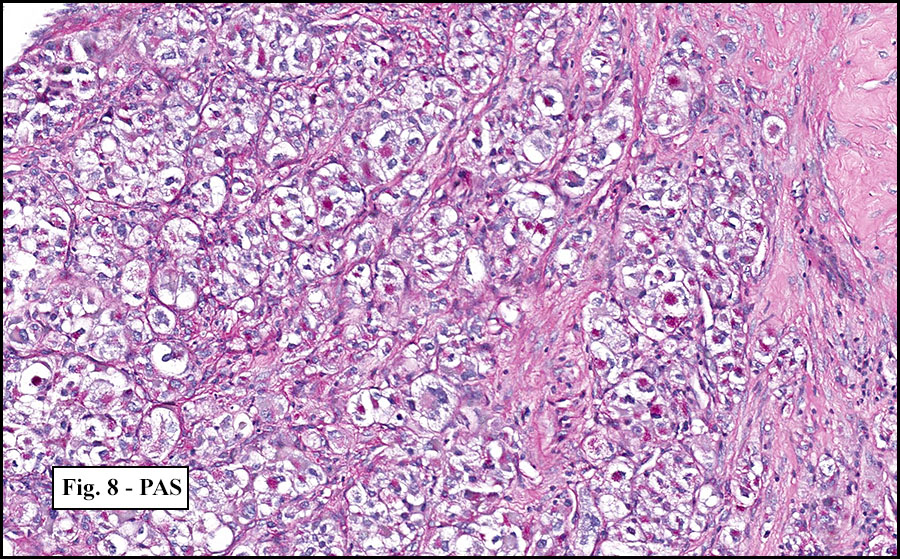
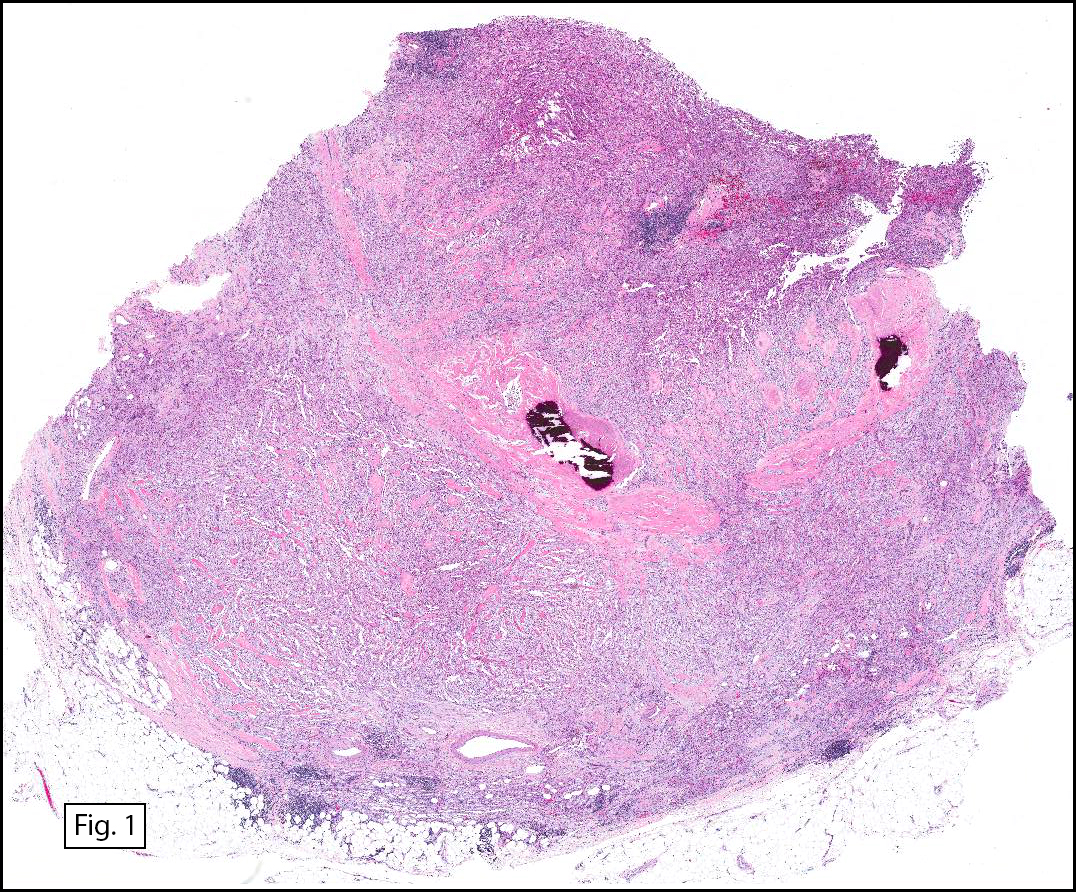
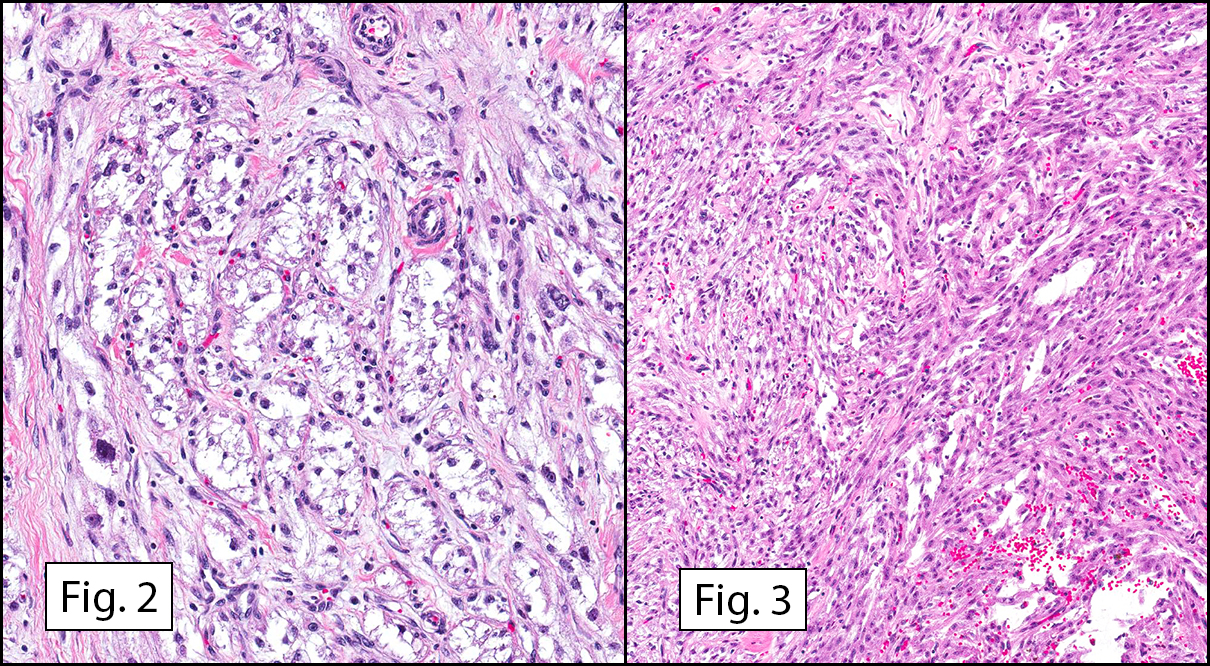
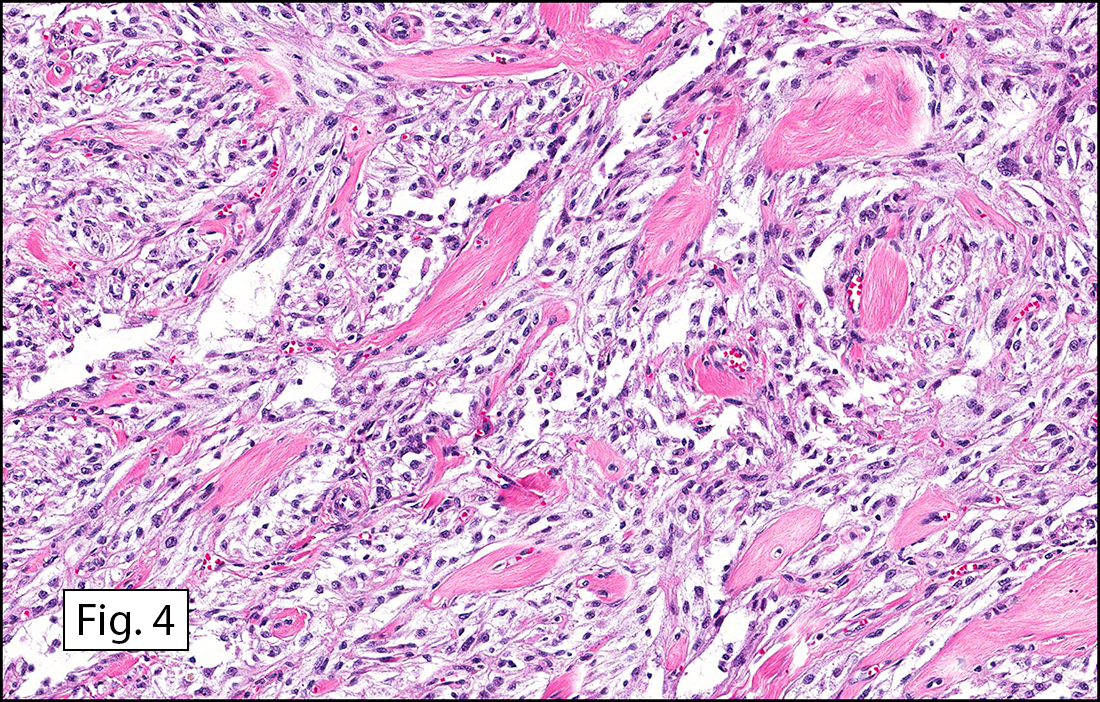
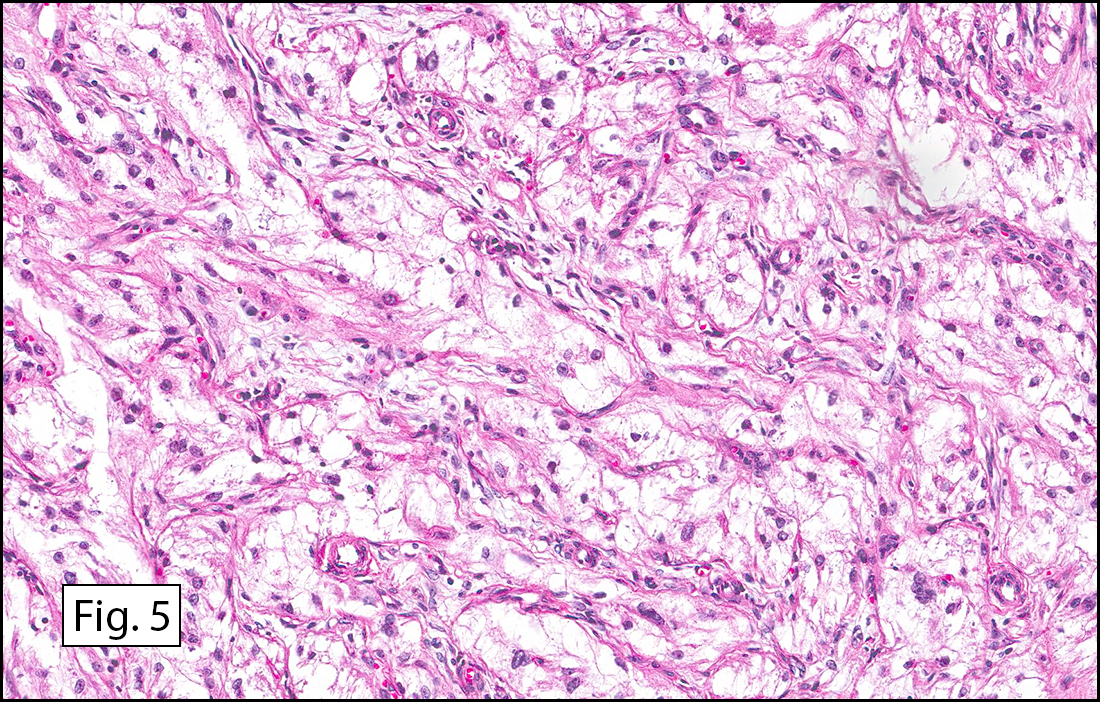
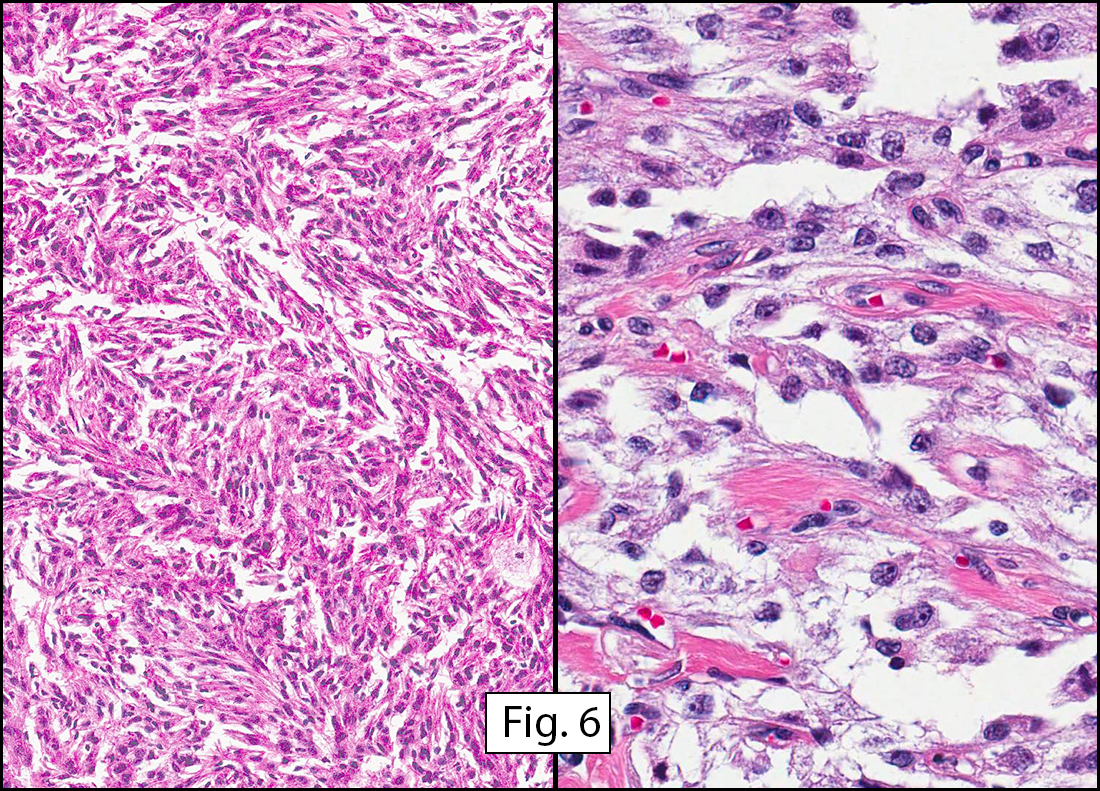
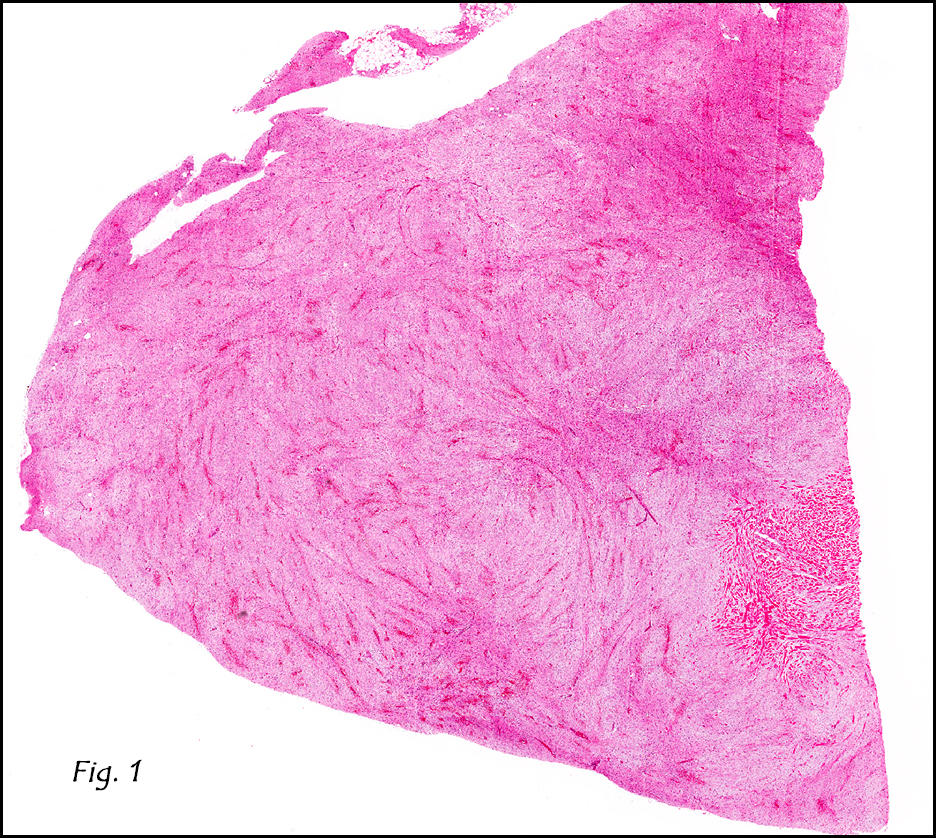
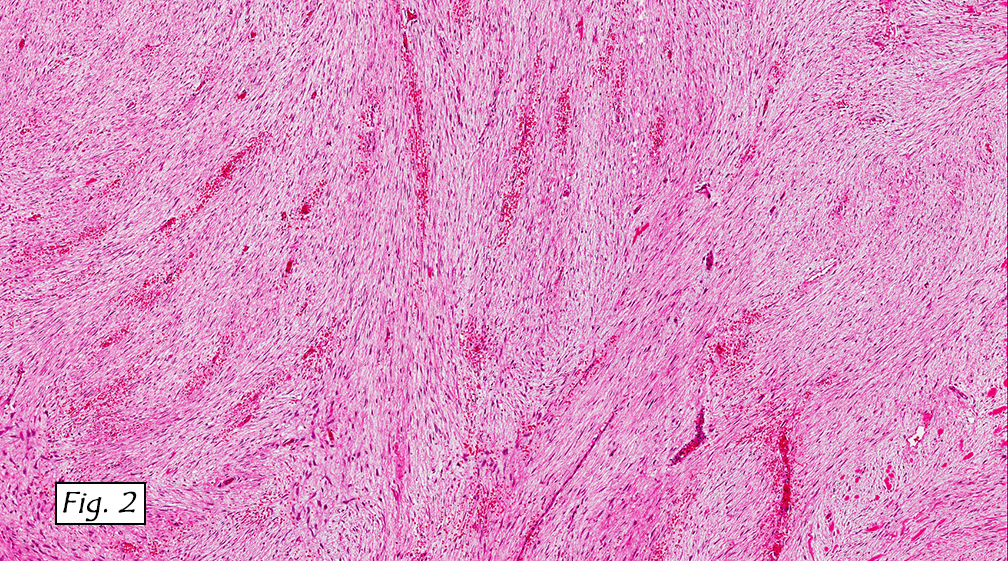
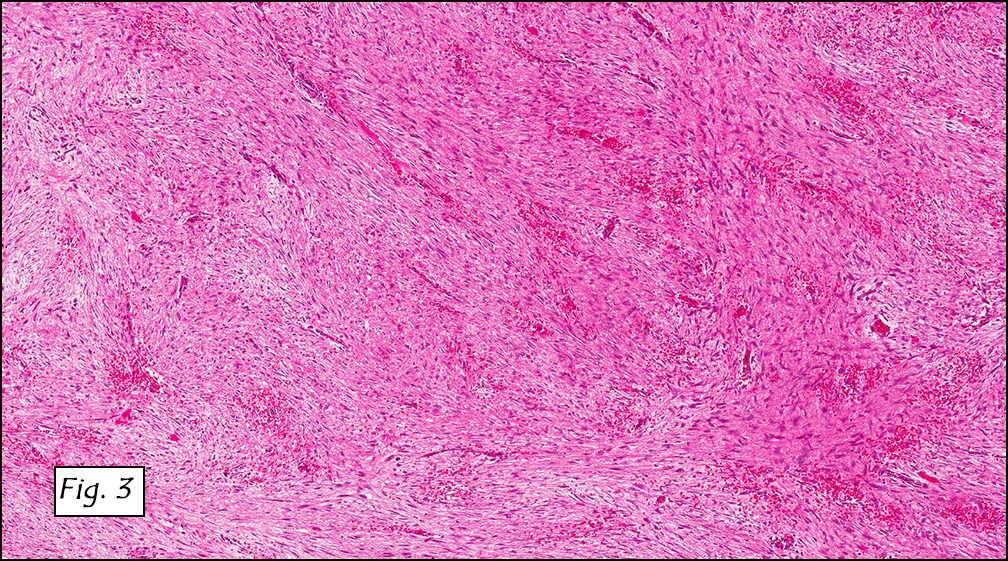
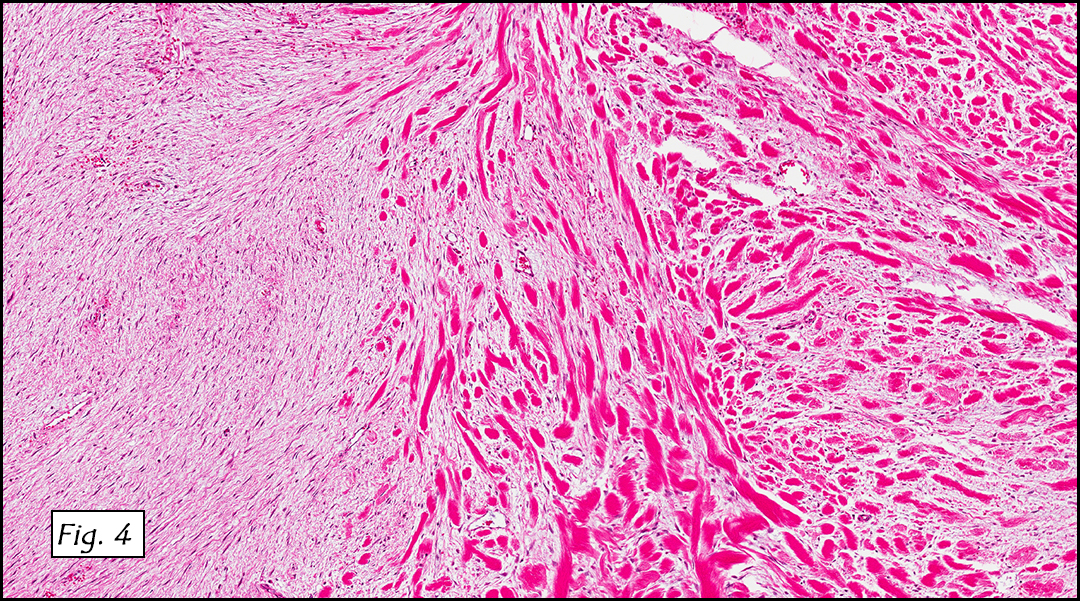
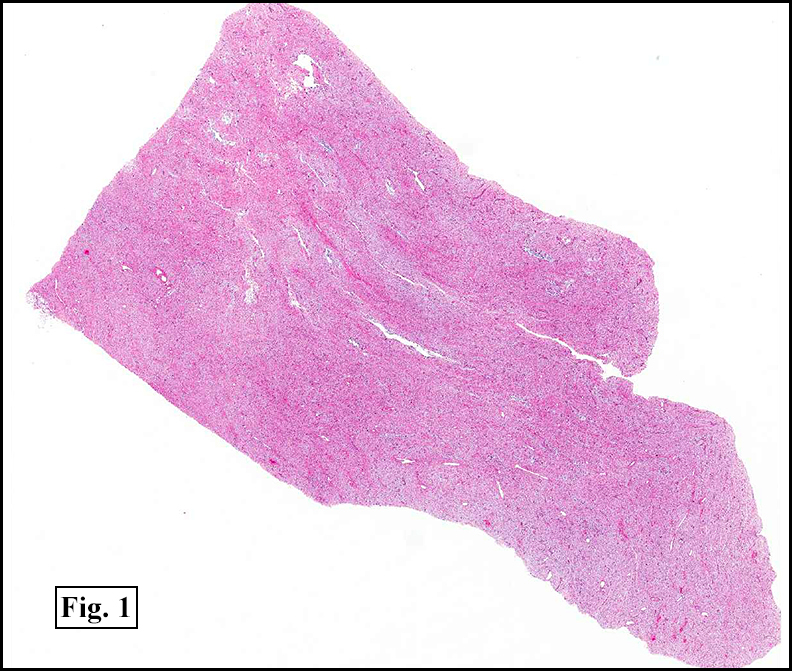
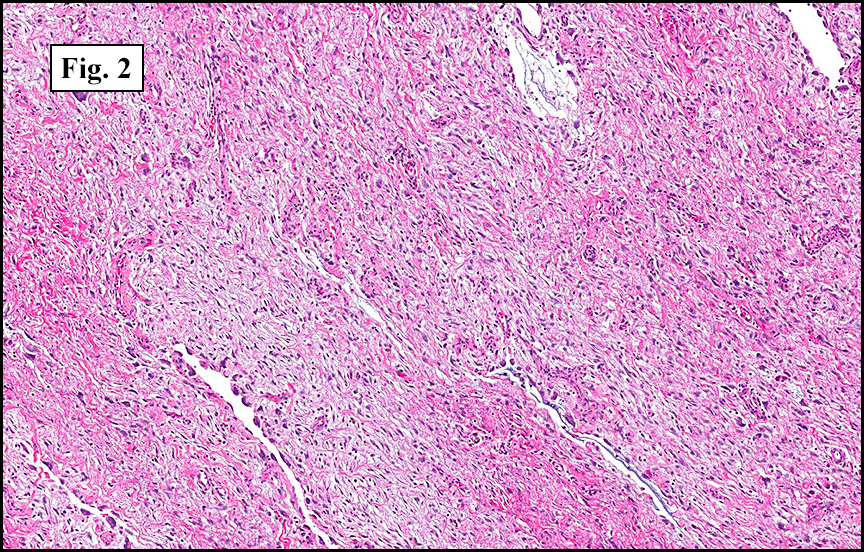
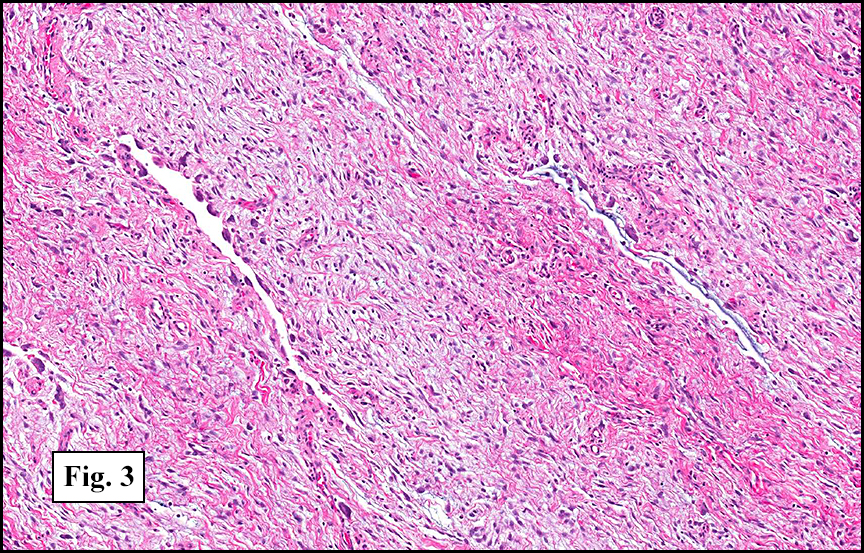
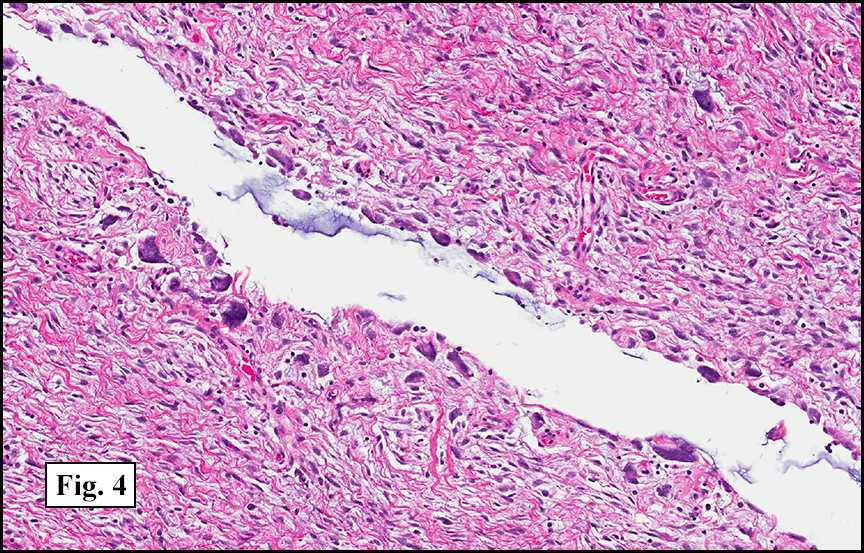
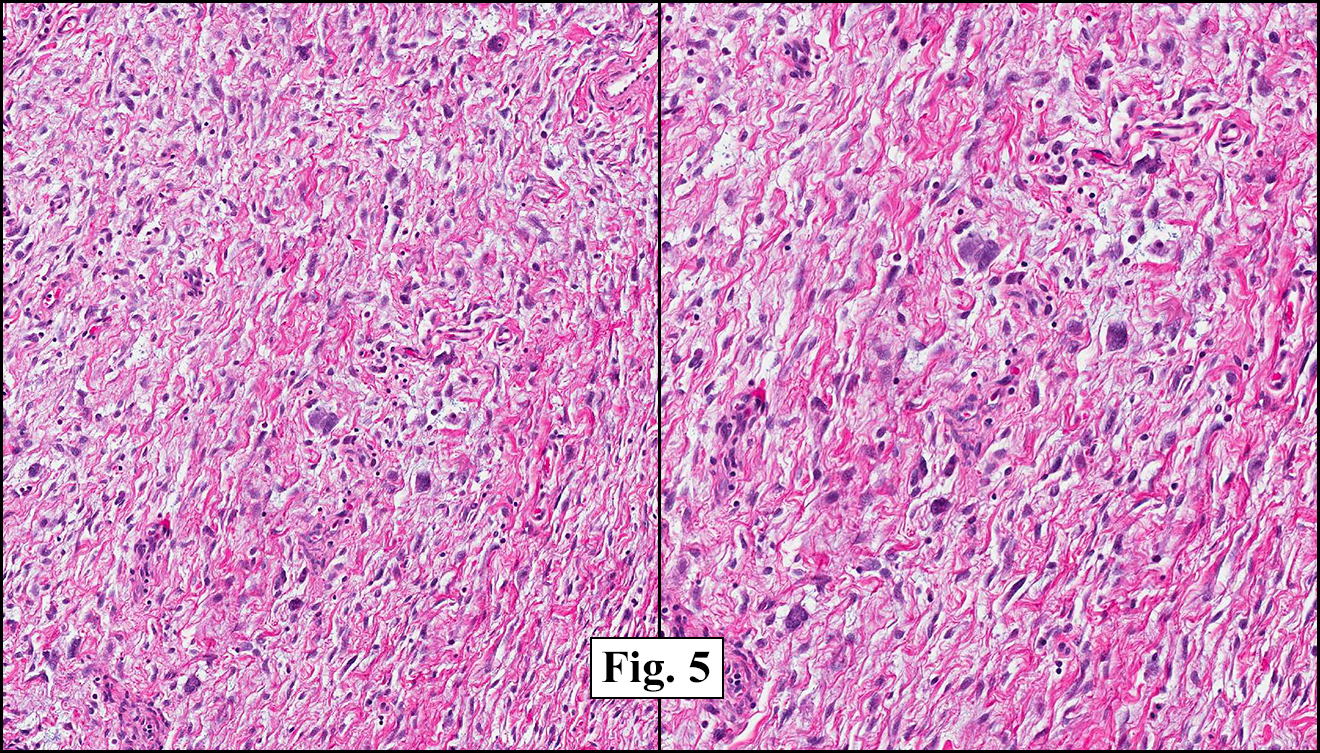
The tumor was compartmentalized, showing islands of cells separated into large and small nodules by fibrous stroma (Figs. 1,2). Smaller nests were bounded by sinusoidal vascular structures, a few of which were dilated (Figs. 3,4). Some of these smaller compartments showed central necrosis and many exhibited clearing around the cells with poor cellular cohesion (Fig. 5). The cells were uniform in both shape and size, and contained abundant pink granular cytoplasm and eccentrically placed, rounded, vesicular nuclei, most having prominent nucleoli (Figs. 6,7). There were scattered binucleated cells (Fig. 7). Mitoses were rare and no significant pleomorphism was present. A PAS stain showed abundant PAS positive granular and crystalloid material within the cytoplasm of many cells (Fig. 8).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Diagnosis: “Alveolar Soft Part Sarcomaâ€
Cody S. Carter, MSIV, Donald R. Chase, M.D.
Department of Pathology and Human Anatomy, Loma Linda University and Medical Center, Loma Linda, California
California Tumor Tissue Registry, Loma Linda, California
Discussion: Alveolar soft part sarcoma (ASPS) is a rare entity of unknown origin accounting for less than 1% of sarcomas. It usually occurs in ages 15-35, and predominantly affects females, especially in younger patients. In adults, the majority of tumors occur in the lower extremities, especially in the anterior thigh and buttocks. In children, these tumors typically present in the head and neck, especially the orbit and tongue, and tend to be smaller, likely due to earlier detection in these locations. The tumor most commonly presents as a slow-growing painless mass, but can often present with respiratory symptoms or neurologic symptoms like headache, visual changes and nausea due to their striking proclivity for early metastasis to the lung and brain. ASPS are typically very vascular and may be associated with an audible bruit. Radiologically, the diagnosis my be suggested byits hypervascularity with prolonged capillary staining and locally dilated veins on angiography and CT scan.
Grossly, ASPS is usually poorly circumscribed and highly vascular, with increased risk of significant blood loss at the time of surgery. The cut surface is usually yellow-white to gray and can feature areas of necrosis or hemorrhage, making the tumor soft and friable.
Microscopically, ASPS shows little variability. Compartments and nests are divided by fibrous stroma, thin-walled sinusoidal channels, usually lined by flattened endothelial cells. A lack of cellular cohesion and central necrosis is often seen. The dominant pattern of peripherally viable tumor cells with partial central clearing likens this neoplasm to the alveoli of the lung. While these features generally make a histologic diagnosis straightforward, the nest-like pattern can, on rare occasion, be absent, and large sheets of cells are seen together. This variant is primarily seen in infants and young children.
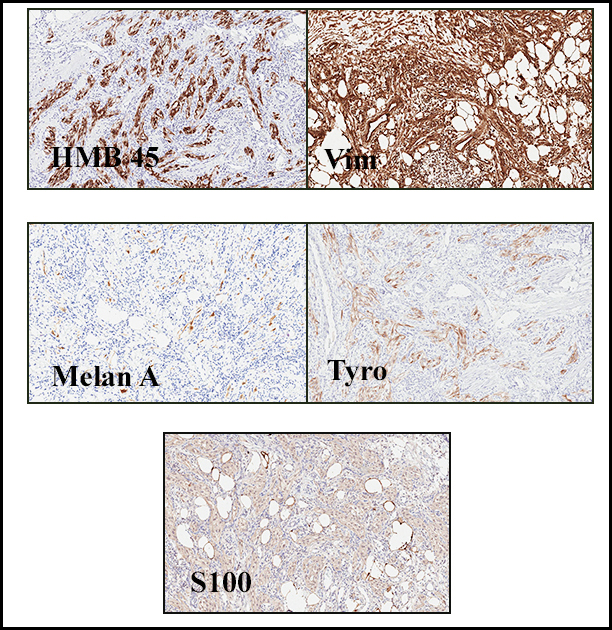
Cells within the nests are large, rounded, and fairly uniform, with eosinophilic granular cytoplasm and vesicular nuclei with distinct nucleoli. Cellular pleomorphism is only rarely present. Vascular invasion is almost invariably present, and reflects the tendency for early blood metastasis. The characteristic histologic pattern can usually be relied on for diagnosis, and the unreliable staining exhibited by ASPS makes immunohistochemistry most important in excluding other neoplasms. Of the histologic stains, PAS will likely show positively staining, diastase-resistant granules or rhomboid and rod-shaped crystals within the cytoplasm. These structures are composed of monocarboxylate tansporter 1 and its chaperone protein CD147. ASPS fails to express cytokeratin, EMA, neurofilament, GFAP, serotonin, synaptophysin and chromogranin. It can sometimes be positive for S-100 and neuron-specific enolase, but these seem to have no significant diagnostic value. Stains for MyoD1 and myogenin have been controversially positive, with one study using positivity in several tumors as support for skeletal muscle differentiation, but several subsequent studies have since failed to reproduce the results.
Cytogenetics may be a useful tool as the unbalanced translocation t(X;17)(p11.2q25) leads to an ASPL-TFE3 fusion gene that is both sensitive and specific for ASPS within the realm of soft tissue sarcomas (a small subset of pediatric renal cell carcinomas have also shown this same fusion gene). This can be more easily confirmed with an immunohistochemical stain for TFE3. This fusion gene has been linked to the overexpression of the promitotic MET receptor tyrosine kinase, which has suggested a model for oncogenesis, as well as a source of targeted therapy that is currently being tested in ongoing clinical trials.
Alveolar soft tissue sarcoma displays indolent growth, but overall prognosis is poor due to early metastasis, which can often be present at initial presentation. ASPS differs from most other adult sarcomas in that local recurrence is less likely after radical excision, but late metastasis can occur years after resection of the primary, and indeed metastases upwards of 30 years later have been recorded. Poorer prognosis is associated with increasing age and increasing tumor size at diagnosis, as well as metastases at initial presentation. Histologic grading is not contributory to prognosis.
Due to slow growth of the tumor, radical surgical excision of both primary and metastatic lesions is the most effective treatment, and conventional radiotherapy and chemotherapy have thus far been shown to be ineffective. Recent advances in targeted therapy are promising, and clinical trials are ongoing.
Differential diagnosis includes:
• Renal cell carcinoma (RCC) can look similar to the histology of ASPS, especially because some variants of the sarcoma can have less eosinophilic cells, and some RCCs can have quite eosinophilic cytoplasm. RCC can be distinguished by its distinct lack of PAS-positive crystalline structures or granules. Also, RCC is immunoreactive to EMA and keratin, but ASPS shows absent staining. TFE3 positivity can also be helpful, but some RCCs can express this antigen in the pediatric population. In this situation, radiologic correlation with renal imaging may be essential.
• Paraganglioma can be differentiated by its positivity for neuroendocrine markers, which are consistently negative in ASPS. Vesicular nuclei and prominent nucleoli are also not characteristics of paraganglioma. Also, paragangliomas typically present in patients over 40 years of age and are not found in the extremities; in contrast, those alveolar soft part sarcomas that arise in the head and neck region are found in pediatric patients.
• Granular cell tumors typically have a solid architecture with cells exhibiting more densely eosinophilic cytoplasm, and can exhibit spindling of the cells, a feature not present in ASPS. Also, cells are uniformly positive for S-100, while ASPS shows variably positivity.
• Alveolar rhabdomyosarcoma (AR) is characterized by smaller cells that are often more pleomorphic and feature dense nuclei. They are also much more likely to mark for desmin, MyoD1, and other muscle markers. The clusters of cells in an AR are surrounded by actual fibrous septa, as opposed to sinusoidal vessels.
Suggested Reading:
Portera CA Jr, Ho V, Patel SR, Hunt KK, Feig BW, Respondek PM, Yasko AW, Benjamin RS, Pollock RE, Pisters PW. Alveolar soft part sarcoma: clinical course and patterns of metastasis in 70 patients treated at a single institution. Cancer. 2001 Feb 1;91(3): 585-91.
Lieberman PH, Brennan MF, Kimmel M, Erlandson RA, Garin-Chesa P, Flehinger BY. Alveolar soft-part sarcoma. A clinico-pathologic study of half a century. Cancer. 1989 Jan 1;63(1):1-13.
Mitton B. Federman N. Alveolar soft part sarcomas: molecular pathogenesis and implications for novel targeted therapies. Sarcoma; 2012:428789. Epub 2012 Apr 8.
Folpe AL, Deyrup AT. Alveolar soft-part sarcoma: a review and update. J Clin Pathol. 2006 Nov;59(11): 1127-32.
Kayton ML, Meyers P, Wexler LH, Gerald WL, LaQuaglia, MP. Clinical presentation, treatment and outcome of alveolar soft part sarcoma in children, adolescents, and young adults. J Ped Surg. 2006 Jan;41(1): 187-93.
M. Ladanyi, M. Y. Lui, C. R. Antonescu et al. The der(17)t(X;17)(p11;q25) of human alveolar soft part sarcoma fuses the TFE3 transcription factor gene to ASPL, a novel gene at 17q25. Oncogene. 2001 Jan 4;20(1):48-57.
Tsuji K, Ishikawa Y, Imamura T. Technique for differentiating alveolar soft part sarcoma from other tumors in paraffin-embedded tissue: comparison of immunohistochemistry for TFE3 and CD147 and of reverse transcription polymerase chain reaction for ASPSCR1-TFE3 fusion transcript. Hum Pathol. 2012 Mar;43(3):356-63.
Weiss S, Goldblum J. Enzinger & Weiss’s Soft Tissue Tumors (5th ed). Philadelphia: Mosby/Esevier Inc. 177-92, 2008.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}