History: A 67-year-old woman presented with a 5 year history of non-tender swelling in the perineal area. There had been no drainage or infection. Physical examination showed a 3 x 2 cm, firm mass attached to the skin just below and lateral to the labia. It was superficial and did not involve underlying tissues.
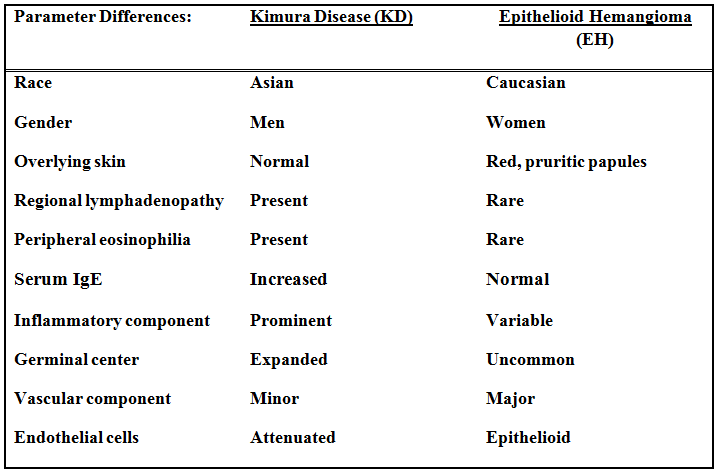
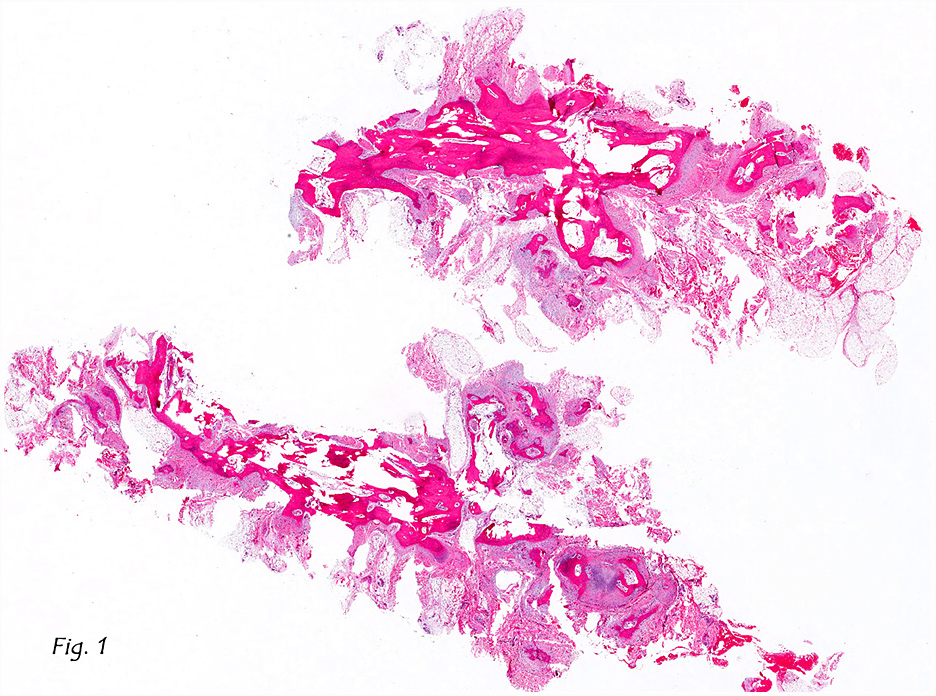
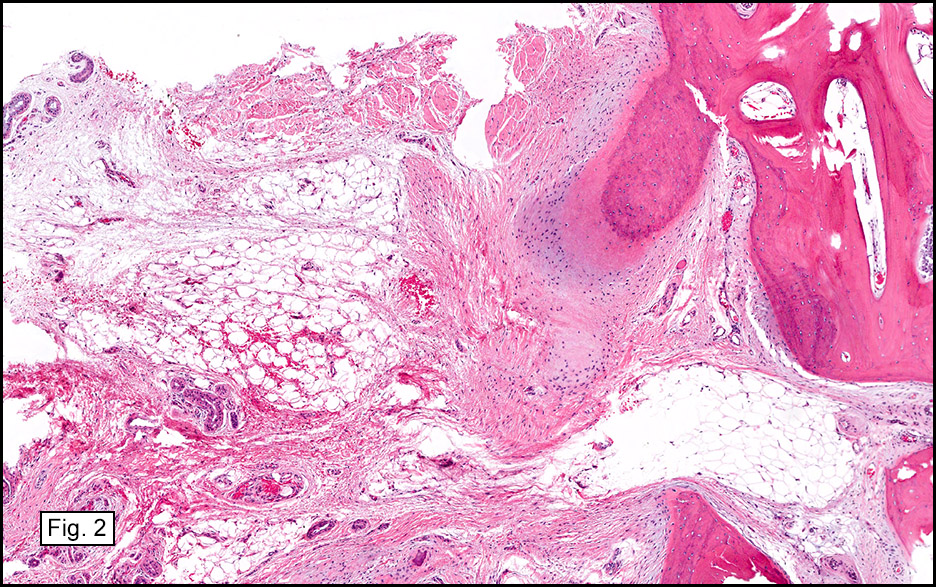
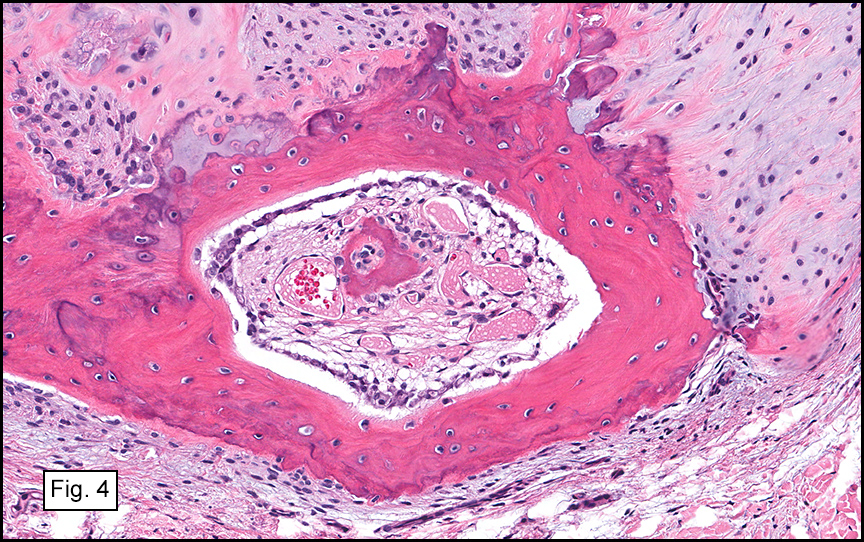
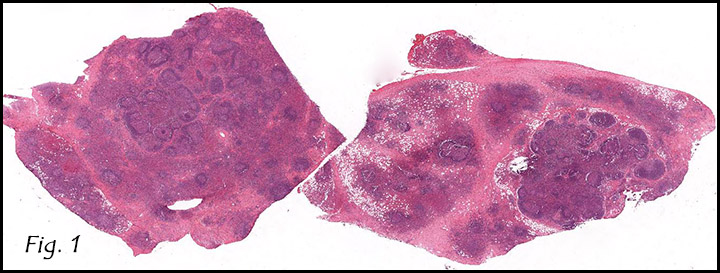
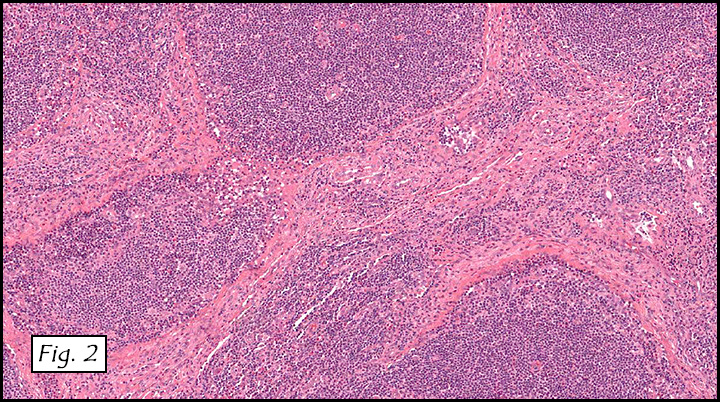
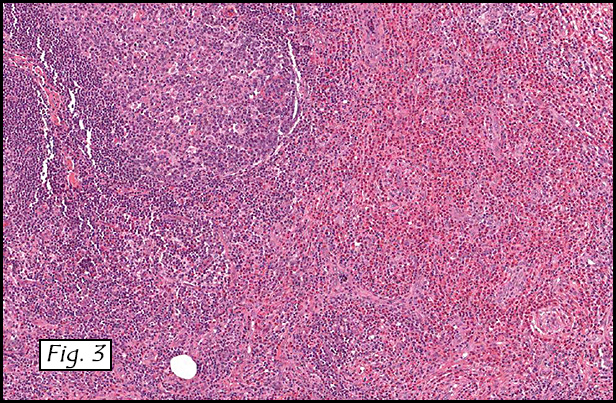
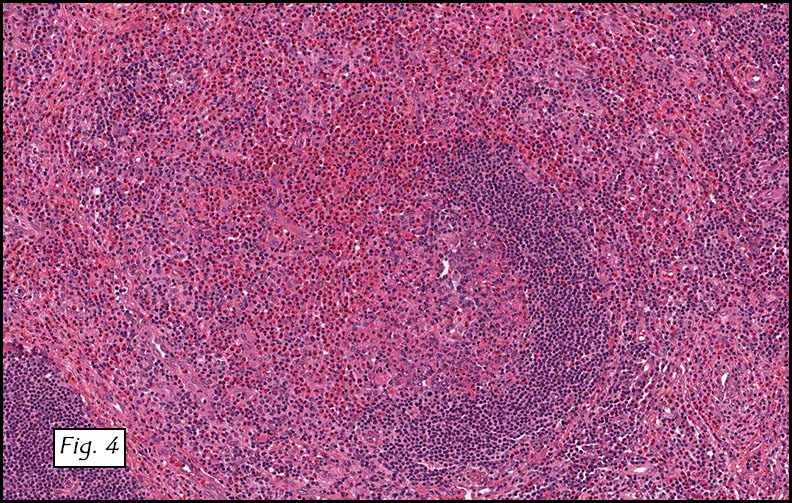
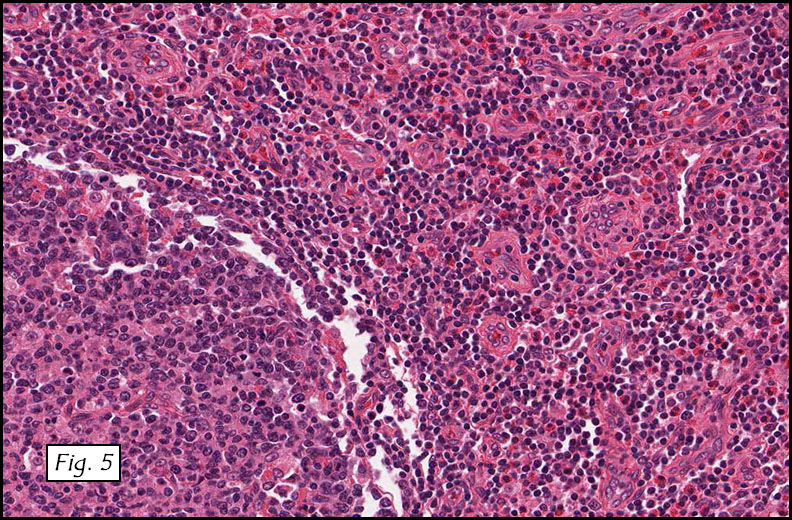
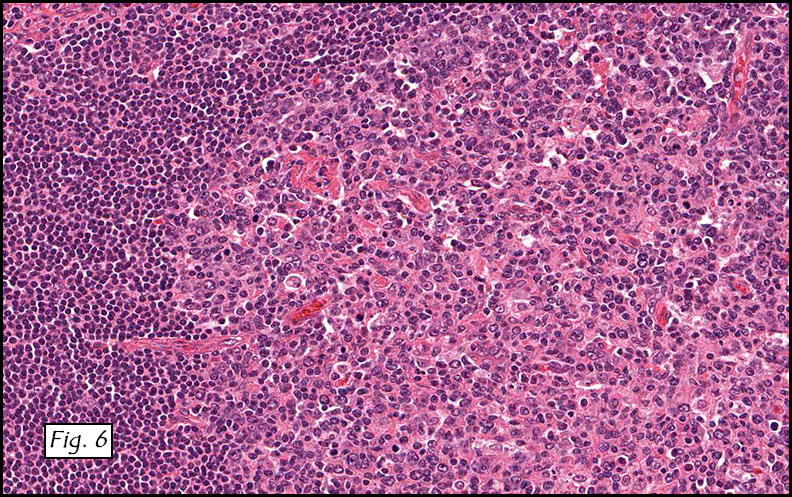
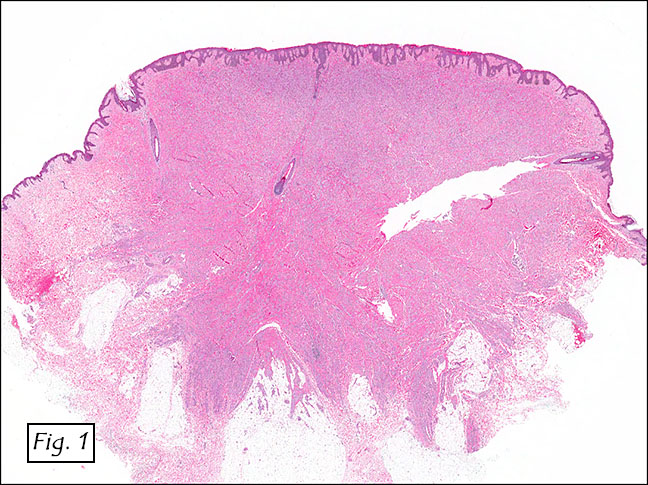
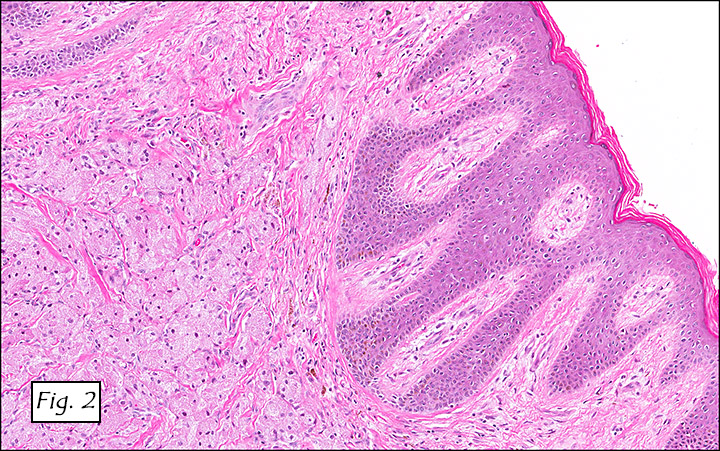
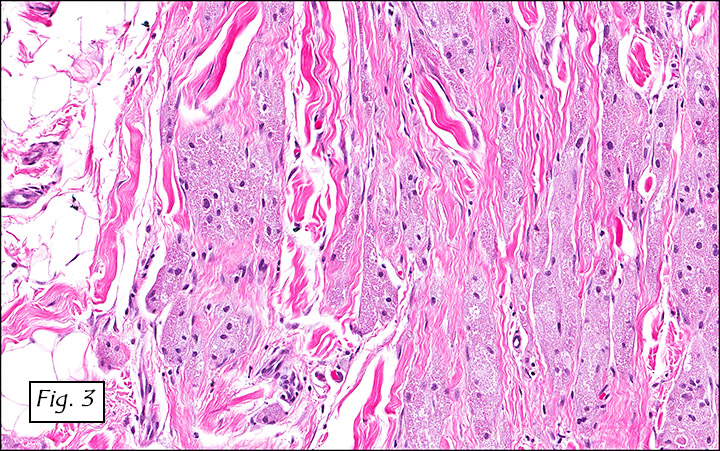
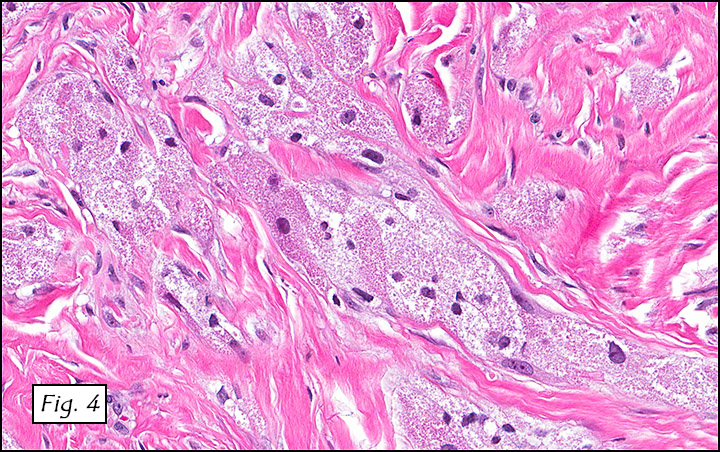
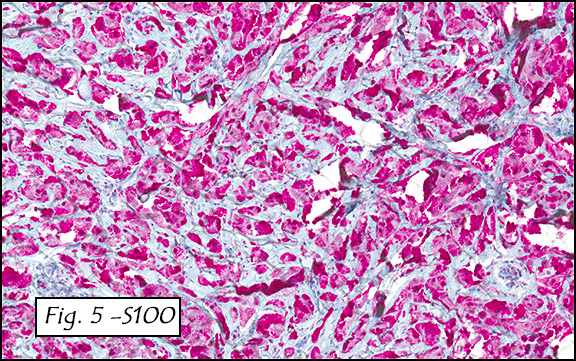
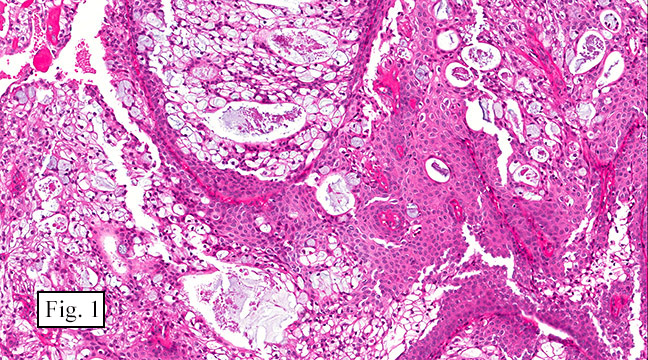
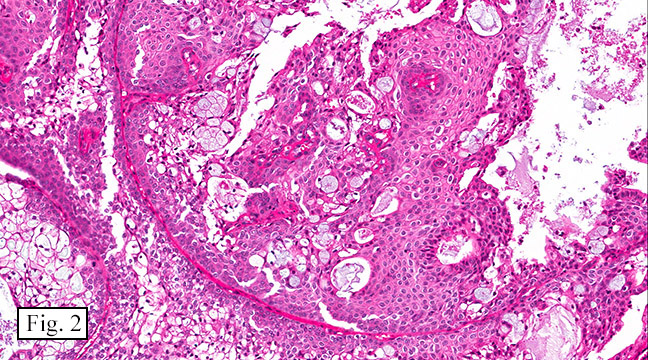
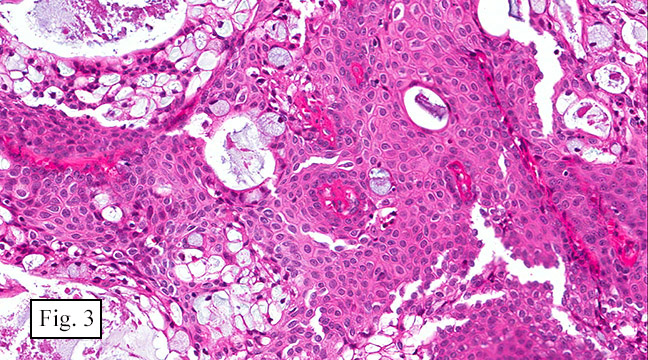
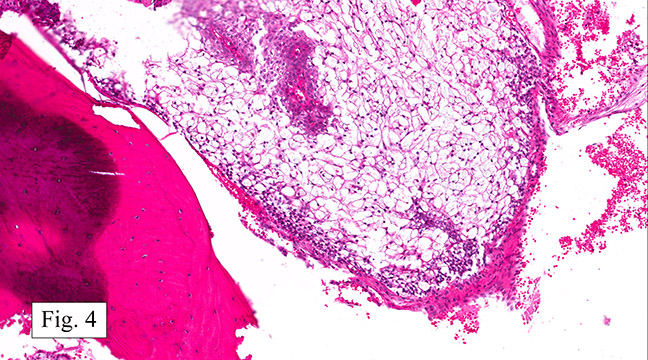
The excised specimen consisted of a yellow-tan, ill-defined, 2.5 x 1.4 x 0.1 cm fibrous subcutaneous nodule (Fig. 1). The overlying epidermis showed elongated hyperplastic rete ridges (Fig. 2). The tumor was composed of sheets of eosinophilic cells with infiltrative borders (Fig. 3). Higher power views demonstrated large polygonal cells with eosinophilic granular cytoplasm, indistinct cell borders, and centrally-situated, small, hyperchromatic nuclei (Fig. 4). No spindling, atypia, pleomorphism, mitoses, nor necrosis were identified. The tumor cells were positive for S-100 (Fig. 5).
Diagnosis: Granular cell tumor, vulva
Li Lei, PGY2 and Donald R. Chase, M.D.
Department of Pathology and Human Anatomy,
Loma Linda University Medical Center, Loma Linda, California
California Tumor Tissue Registry, Loma Linda, California
Discussion: Granular cell tumor (GCT) was first described by Virchow and Weber in 1854, and was later characterized as granular cell myoblastoma by Abrikossoff in 1926. Other names, such as Abrikossoff’s tumor, and granular cell schwannoma, have also been used. After decades of debate regarding the histogenesis, most believe that GCT is of neurogenic origin, most likely Schwann cells. Others advocate that GCT is a degenerative change resulting in a cytoplasmic accumulation of lysosomes that occur primarily in Schwann cells as well as in a variety of other cell types. Despite the unresolved controversy on the histogenesis, the simple term “GCT†is currently used by most pathologists.
GCT accounts for 0.5 – 1.3% of all soft tissue tumors. It occurs in patients of all age groups, with a peak incidence in the third to sixth decades. Women are affected twice as often as men. GCT can occur anywhere in the body, but most commonly involves the tongue. It typically presents as a solitary, usually asymptomatic nodule, though pruritus, pain, and ulceration have occasionally been reported. GCT involving critical anatomic sites such as trachea and neurohypophysis can cause serious complications. Between 3 to 20% of patients have multiple tumors that appear synchronously or metachronously, making it a challenge in some cases to determine if the tumor is primary or metastatic.. Multiple GCTs may be part of LEOPARD syndrome which is associated with PTPN11 mutation. Rarely, familial cases have been reported, suggesting that GCT may be inheritable.
Approximately 5 – 16% of GCTs occur in the vulvar region. For reasons unknown, it is more common in African-Americans than Caucasians. Vulvar GCT generally involves the labium majus, though clitoris, mons pubis, perineum and perianal region may also be affected. The male counterparts, GCTs of scrotum and/or penis, are extremely rare with only 22 cases reported in the literature. Vulvar GCT is usually slow-growing, but may enlarge rapidly during pregnancy. Although most vulvar mesenchymal tumors express estrogen receptors and/or progesterone receptors, it is still unclear if vulvar GCTs do.
An MRI may help to diagnose the tumor pre-operatively. GCT usually shows a peripheral hyperintense rim encircling a central hypointense core. These features are most appreciated on T1-weighted images, and may become more prominent on T2-weighted images. There is also variable peripheral enhancement in post-contrast images.
Grossly, GCT is a stellate-shaped or circumscribed, nonencapsulated, subcutaneous mass. The cut surface is tan to yellow, firm and homogeneous. It is usually less than 3 cm, but may grow up to 12 cm.
Microscopically, GCT displays either nodular or infiltrative growth pattern(s). It is composed of sheets, nests, or cords of large polygonal cells, which are characterized by indistinct cytoplasmic borders, abundant eosinophilic granular cytoplasm, and centrally-located small oval nuclei with hyperchromasia. Within the fine or coarsely granular cytoplasm, larger eosinophilic globules surrounded by clear halos are sometimes seen.
Benign GCTs may be slightly spindled, however prominent spindling is a clue of more aggressive behavior. Fibrosis or desmoplasia is variable. An important but nonspecific feature of these tumors is pseudoepitheliomatous hyperplasia of overlying squamous epithelium. This can be mistaken for well-differentiated squamous cell carcinoma if the tumor is only superficially sampled.
The cytoplasmic granules of tumor cells may contain large amounts of hydrolytic enzymes, which are positive for Luxol fast blue and Periodic Acid-Schiff (PAS) with diastase resistance. The cells show diffuse strong nuclear and cytoplasmic S-100 positivity, though some malignant GCTs may lose S-100 expression. Other ancillary positive markers include neuron specific enolase, myelin basic protein, laminin, calretinin, vimentin, osteopontin, inhibin-alpha, HLA-DR, protein gene product 9.5, CD68, carcinoembryonic antigen (CEA), and CD57 (Leu-7). The tumor cells are negative for cytokeratin, desmin, chromogranin, HMB-45, and glial fibrillary acidic protein (GFAP).
Ultrastructurally, the tumor cells are surrounded by replicated basal lamia suggestive of repeated cycles of cellular injury and repair. This is another feature supporting the degenerative theory of histiogenesis. The cytoplasmic granules are composed of membrane-bound, autophagic, vacuoles containing myelin figures and fragmented rough endoplasmic reticulum and mitochondria, consistent with phagolysosomes. There are also small interstitial cells with angulated bodies resulting in a Gaucher cell-like appearance.
Approximately 98% of the GCTs are benign. However, malignant transformation can develop as soon as 5 months after an initial benign diagnosis. Features suggestive of malignancy include large size (>4 cm), rapid growth, necrosis, spindling of tumor cells, vesicular nuclei with prominent nucleoli, high Ki67 labeling index (>10%), > 2 mitoses/10 high power fields at 200X magnification, high nuclear to cytoplasmic ratio, and pleomorphism. Note that mild to moderate nuclear atypia is not indicative of malignancy. Occasionally, granular cells may be found in in the wall or lumen of supporting blood vessels, but this phenomenon does not portend aggressive behavior. Malignant GCTs are usually very aggressive with local recurrence rates up to 70% and 3-year survival rates of less than 50%. They often metastasize to regional lymph nodes, lungs, liver, and bones. Since metastases may occur more than 10 years after treatment, long-term follow-up is necessary.
The differential diagnosis includes:
• Reactive histiocytic proliferation that occurs as nodular collections of histiocytes in response to a variety of stimuli such as trauma or foreign materials. The histiocytes typically have reniform vesicular nuclei, and are arranged to circumscribe the necrotic debris. A detailed clinical history also helps in making the correct diagnosis.
• Genital rhabdomyoma usually grows as a polypoid mass in younger patients. Tumor cells are characterized by strap-like or epithelioid morphology, cytoplasmic cross striations, large cigar-shaped nuclei, and immunoreactivity for muscle markers such as desmin.
• Genital leiomyoma usually shows myxoid change and an epithelioid phenotype. The tumor cells are positive for desmin and smooth muscle actin, and negative for S-100.
• Hibernoma also shows granular eosinophilic cells and strong S-100 positivity. The more unique features are multivacuolated cytoplasm with multiple small lipid droplets, which can be demonstrated by lipid stain such as Oil Red O. Moreover, vulvar hibernoma is exceedingly rare with only one case reported in the literature.
• Squamous cell carcinoma rarely has granular cells. In younger patients it is usually related to human papilloma virus (HPV) infection, and is positive for p16.
• Alveolar soft part sarcoma can be confused with malignant GCT on H&E. It is most commonly seen in patients 15 to 35 years of age but rarely involves the vulva. PAS staining with diastase reveals cytoplasmic glycogen and rhomboid or rod-shaped crystals. Most of cases show nuclear immunoreactivity for transcription factor E3 (TFE3).
• Melanoma may mimic malignant GCT if it is not pigmented. Both show strong nuclear and cytoplasmic S-100 staining. The diagnosis of melanoma can be confirmed by HMB-45 and Melan-A positivity as well as identification of melanosomes and premelanosomes on electron microscopy.
Treatment depends on the biologic behavior and growth pattern of the tumor. Benign GCTs with well-defined pushing borders are usually cured by simple local excision. Those with infiltrative borders need wide local excision to minimize the risk of recurrence. Some of them still recur even with negative surgical margins, and this may be the first sign of potential aggressive behavior. Malignant GCT warrants more aggressive intervention including radical local surgery with or without regional lymph node dissection. The role of radiation is controversial, however it seems that a dose of 60 Gy or higher may be necessary to be effective. The tumor does not respond to chemotherapy.
Suggested Reading:
Goldblum J, Folpe A, Weiss S. Enzinger & Weiss’ Soft Tissue Tumors, 6th ed: Philadelphia, Elsevier Inc, 2014; 43-44, 838-845.
Rosai J. Rosai and Ackerman’s Surgical Pathology, 10th ed: Philadelphia, Elsevier Inc, 2011; 2181-2182.
Abdullgaffar B, Keloth TR, Raman LG, Mahmood S, Almulla A, Almarzouqi M, Al-Hasani S. Unusual benign polypoid and papular neoplasms and tumor-like lesions of the vulva. Ann Diagn Pathol. 2014; 18(2):63-70.
Tawfiq N, Sabri S, Saiss K, Bouchbika Z, Benchekroun N, Jouhadi H, Sahraoui S, Benider A. Granular cell tumor: report of a complicated vulvar localization of pulmonary metastases. Cancer Radiother. 2013; 17(7):671-674.
Ramos PC1, Kapp DS, Longacre TA, Teng NN. Malignant granular cell tumor of the vulva in a 17-year-old: Case report and literature review. Int J Gynecol Cancer. 2000; 10(5):429-434.
Fanburg-Smith JC, Meis-Kindblom JM, Fante R, Kindblom LG. Malignant granular cell tumor of soft-tissue: diagnostic criteria and clinicopathologic correlation. Am J Surg Pathol 1998; 22:779-94.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}