History: A healthy Caucasian girl was delivered vaginally at full term, with no complications. At two weeks, a soft, non-discrete, non-tender right cheek mass was noted. The mass was non-pulsatile, did not bleed or interfere with feeding or respiration, nor did the size of the mass fluctuate with crying or movement. There were no abnormalities of the overlying skin or mucosa. The mass was closely observed for the next few months. Ultrasound demonstrated a 4 x 3 x 1.4 cm solid whorled mass anterior to the parotid; MRI revealed a well-circumscribed mass involving the right masseter muscle, adjacent to the mandible with increased T2-signal within the bone marrow of the mandible. At five months of age, she underwent intraoral excision of the mass under anesthesia. The patient, now 16 months old, recovered from surgery without complications. Post-operative MRI demonstrated a residual and/or recurrent 3 cm right masseter mass with no other nodules or masses have been observed. No family history of similar lesions or syndromic conditions was elicited.
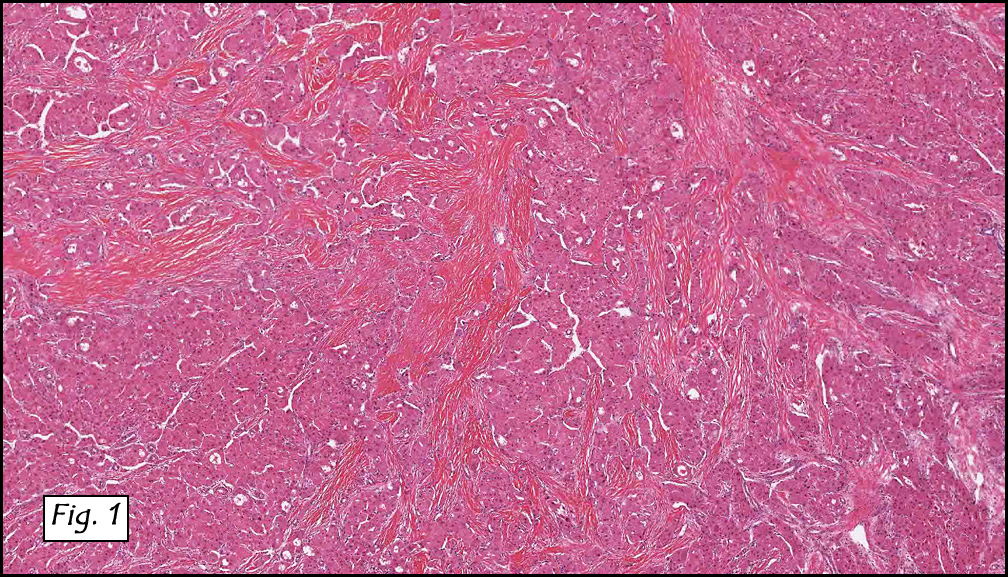
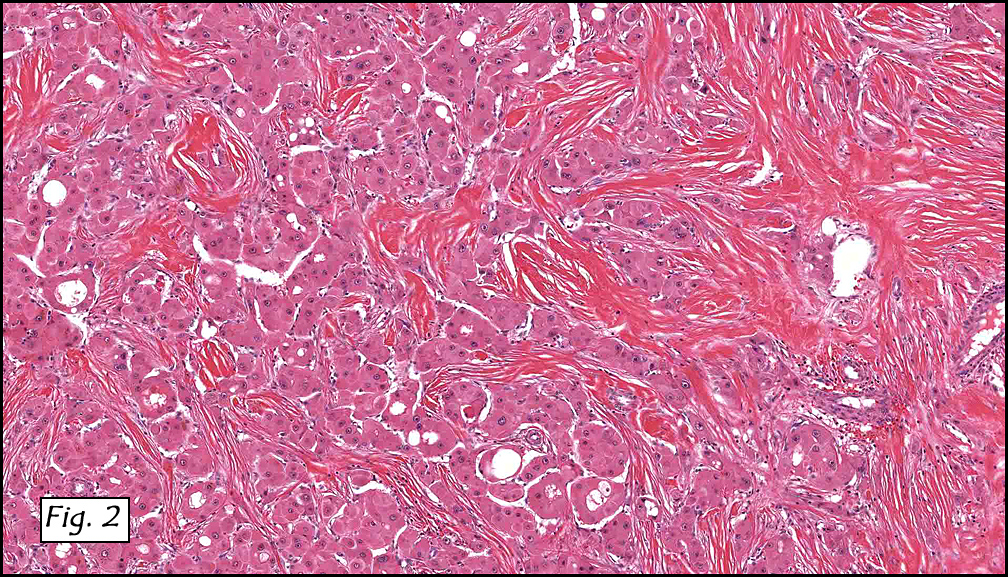
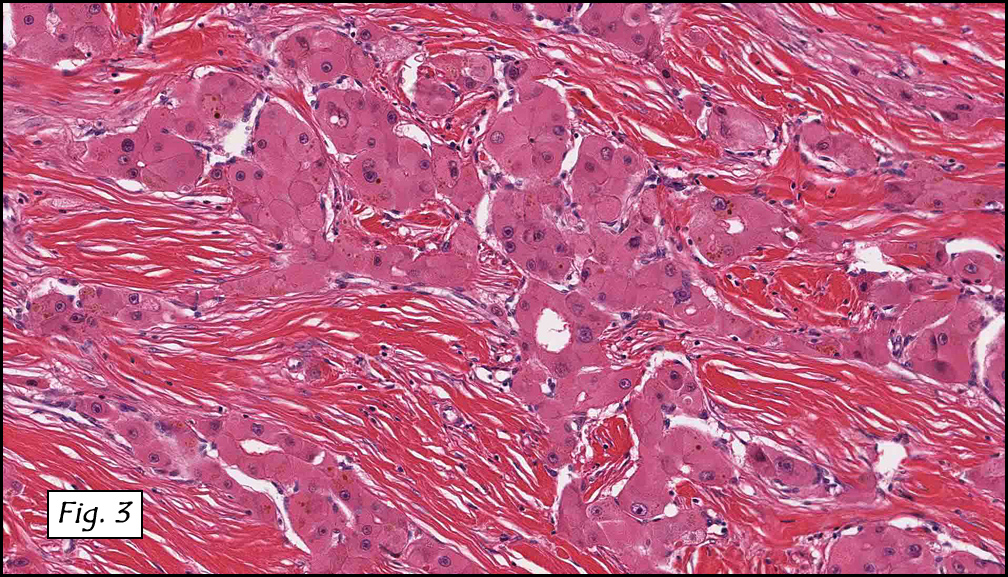
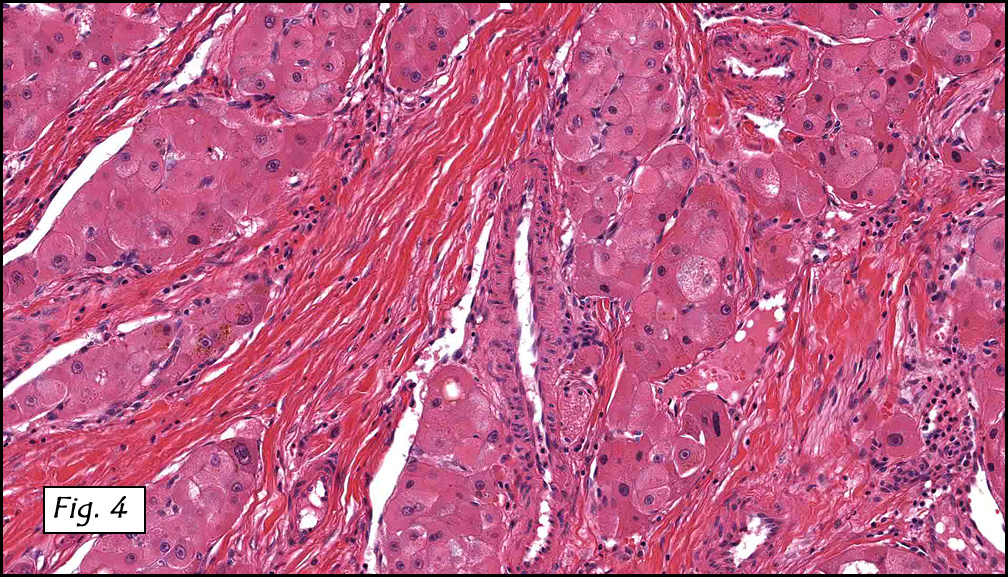
Resection of the lesion yielded an aggregate of approximately 2.0 x 1.5 x 0.4 cm homogenous brown soft tissue. Microscopically, the lesion appeared to be well-circumscribed but extended to the inked margins (Fig. 1). Skeletal muscle differentiation and maturation were observed with cross striations and haphazard arrangement of irregular fascicular bundles of muscle cells with scant to moderate eosinophilic cytoplasm (2,3,4). No definite immature cells were identified, and no myxoid stroma was noted. No areas of hypercellularity, increased mitotic activity, pleomorphism or necrosis were observed.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Diagnosis: “Fetal rhabdomyoma, intermediate (cellular) type, cheekâ€
Evelyn Choo MD1, Rachel Conrad MD1, Anwar Raza, MD1, Donald Chase MD1,2
1. Department of Pathology and Human Anatomy, Loma Linda University and
Loma Linda Medical Center, Loma Linda, California
2. California Tumor Tissue Registry, Loma Linda, California
Discussion: Fetal rhabdomyoma is a rare but benign skeletal muscle neoplasm, first described by Dehner in 1972. It typically occurs in the head and neck region of young children and demonstrates varying degrees of skeletal muscle differentiation.
The term rhabdomyoma encompasses a complex classification system of several distinct benign skeletal muscle neoplasms. It is organized by location (cardiac versus extracardiac), and the extracardiac type is divided into adult, genital and fetal categories based on tissue differentiation and clinical presentation. The adult form tends to occur in head and neck region of 40-60 year old males and displays prominent eosinophilic polygonal cells with vacuolated cytoplasm. The genital form typically presents as a polypoid lesion in vulva and vagina of middle-aged women; microscopically, long strap-like muscle fibers with prominent cross-striations are seen in a collagenous and myxoid matrix. The fetal form shows immature skeletal muscle differentiation and is more common in the head and neck region of children below age four.
The fetal form is further subdivided into a myxoid type and an intermediate type. The myxoid subtype (also known as “classicâ€) is comprised almost entirely of immature primitive spindle cells in a myxoid stroma and tends to appear in postauricular soft tissue. In contrast, a wider spectrum of myocyte maturation is seen in the intermediate subtype (also known as “cellular†or “juvenileâ€), initially described by Di Sant’Agnese and Knowles in 1980. This subtype of rhabdomyoma appears more often in soft tissue of face or mucosal tissue and represents a more differentiated lesion than classic fetal subtype.
Clinically, fetal rhabdomyoma presents in children less than four years old, may often be congenital and is more common in males than females (2.4:1). The lesion consists of a solitary, well-defined, non-tender mass involving soft tissue or mucosa. It grows very slowly and seldom ulcerates the overlying skin or mucosa. Multiple extracardiac rhabdomyomata have been associated with nevoid basal cell carcinoma syndrome (also known as Gorlin-Goltz syndrome) and may show a mutation in PTCH on chromosome 9q22. However, no correlation is seen with tuberous sclerosis as in cardiac rhabdomyoma.
The gross appearance of fetal rhabdomyoma consists of a well-circumscribed, smooth, polypoid mucosal lesion, typically two to six centimeters in diameter. Lesions are typically solitary. Cross-sectioning reveals a grey to pink surface, often with a myxoid appearance.
Microscopically, these lesions display either a myxoid or an intermediate pattern. The myxoid version is comprised of a myxoid matrix surrounding scattered or short bundles of immature spindle-shaped or oval cells with immature skeletal muscle fibers. The small uniform nuclei contain delicate chromatin and are enveloped by tapered eosinophilic cytoplasmic processes. Rare cross-striations are present. The intermediate type closely resembles classic adult head and neck rhabdomyoma, and contains a wider spectrum of myocyte differentiation with few immature spindle-shaped muscle precursors and numerous prominent strap-shaped muscle cells. The myocytes have central vesicular nuclei, scant to moderate eosinophilic cytoplasm, and frequent cross-striations with occasional vacuolation and glycogen. They are haphazardly arranged in irregular fascicular bundles. Ganglion-like rhabdomyoblasts with prominent nucleoli may be present. In both myxoid and intermediate types, the absence of significant atypia, necrosis, or mitotic activity is noted. Invasion and destruction of surrounding tissues should not be seen.
Immunohistochemical staining shows positivity for muscle-specific actin (MSA), myoglobin, and desmin; focal positivity may be seen for smooth muscle actin (SMA), S100, glial fibrillary acidic protein (GFAP), and vimentin.
Electron microscopy will show thick and thin myofilaments in the more mature myocytes; z-bands and glycogen may also be demonstrated in the cytoplasm. The immature myocytes may lack specific ultrastructural features of myocyte differentiation.
The recommended therapy for fetal rhabdomyoma is complete excision. Prognosis is excellent. Local recurrence is rare and is typically associated with incomplete resection. Metastases have never been described in the literature. Only two controversial cases of possible malignant transformation have been reported but may have been misdiagnosed as benign initially, and recurrent cases should be carefully evaluated for a misdiagnosis of rhabdomyosarcoma.
The differential diagnosis of fetal rhabdomyoma includes rhabdomyosarcoma (both embryonal and spindle cell types), adult rhabdomyoma, genital rhabdomyoma, Triton tumor (neuromuscular hamartoma), rhabdomyomatous mesenchymal hamartoma of the skin, and infantile fibromatosis.
1. Rhabdomyosarcoma consists of pronounced diffuse cellular immaturity, necrosis, mitotic figures, and infiltration of adjacent tissues with frank destruction. The presence of nuclear atypia in rhabdomyosarcoma is the most important criteria to distinguish it from fetal rhabdomyoma.
2. Adult rhabdomyoma has distinctive polygonal “spider†cells with vesicular nuclei, central nucleoli, and eosinophilic cytoplasm. It tends to occur in the head and neck region of adult males.
3. Genital rhabdomyoma demonstrates predominately mature long, strap-like striated muscle cells and occasional immature myocytes in a background of myxoid material and collagen. It typically presents as an asymptomatic, slow-growing, small polypoid lesion in the vulvo-vaginal region of middle-aged women. Rare occurrences in the male urogenital tract have also been described.
4. Triton tumor is composed of nerve fascicles mixed with mature striated skeletal muscle. It is associated with neurofibromatosis in younger patients and typically affects the axial skeleton.
5. Rhabdomyomatous mesenchymal hamartoma consists of normal dermal elements with mature striated skeletal muscle.
6. Infantile fibromatosis is not as well-circumscribed as fetal rhabdomyoma. It tends to involve regions deeper than the subcutis, has a fasciculated spindle cell pattern, lacks cytoplasmic striations, and contains interspersed fat cells.
Fetal rhabdomyoma is a rare neoplasm of immature skeletal muscle. Distinguishing it from the more common, more aggressive rhabdomyosarcoma is important. Clinical characteristics (location, behavior) and microscopic traits (mitoses, necrosis, and especially nuclear atypia) may be helpful. Prognosis is excellent after surgical excision. Recognition of this benign neoplasm is important in communicating accurate prognostic information and in preventing over-aggressive treatment.
Suggested Reading:
1. Dehner LP, Enzinger FM, Font RL. Fetal rhabdomyoma: an analysis of nine cases. Cancer. 1972;30:160-166.
2. Di Sant’Agnese PA, Knowles DN. Extracardiac rhabdomyoma: a clinicopathologic study and review of the literature. Cancer. 1980;56:780-789.
3. Kapadia SB, Barr FG. Rhabdomyoma. In: Fletcher CDM, Unni K, Mertens F, eds. World Health Organization Classification of Tumours. Pathology and Genetics of Tumors of Soft Tissue and Bone. Lyon, France: IARC Press, 2002:142-145.
4. Premalata CS, Kumar RV, Saleem KM, Fathima LJ, Das K. Fetal rhabdomyoma of the lower extremity. Pediatr Blood Cancer. 2009;52(7):881-883.
5. Walsh SN, Hurt MA. Cutaneous fetal rhabdomyoma: a case report and historical review of the literature. Am J Surg Pathol. 2008;32(3):485-491.
6. Weiss SW, Goldblum JR. Enzinger & Weiss’s Soft Tissue Tumors. 5th ed. Philadelphia: Mosby-Elsevier, 2008:583-592.
7. Yang S, Zhao C, Zhang Y, Liao S. Mediastinal fetal rhabdomyoma in nevoid basal cell carcinoma syndrome: a case report and review of the literature. Virchows Arch. 2011; 459:235-238.