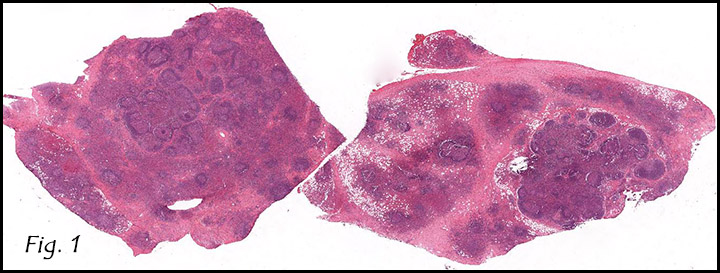
History: A 32-year-old man presented with a mass in the right arm. It involved the humerus and upon excision was 8.2 x 6.5 x 3.5 cm and weighed 52 grams. It consisted mostly of fatty tissue but had a 3.0 x 2.5 cm, vaguely circumscribed nodule centrally located.
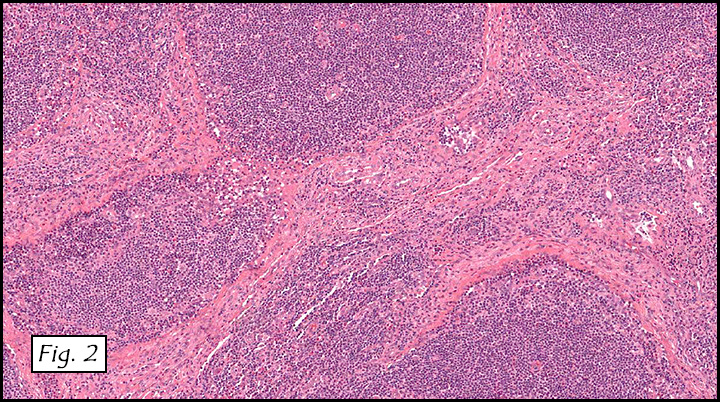
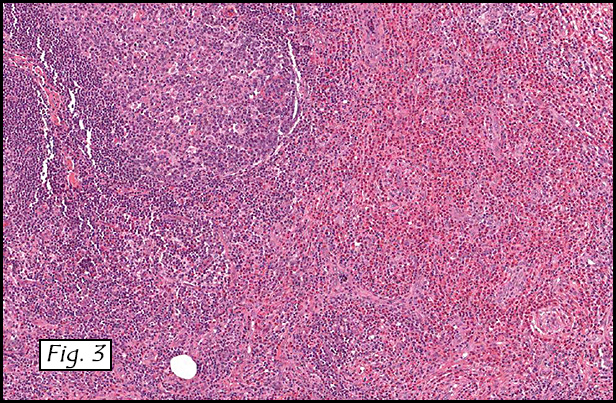
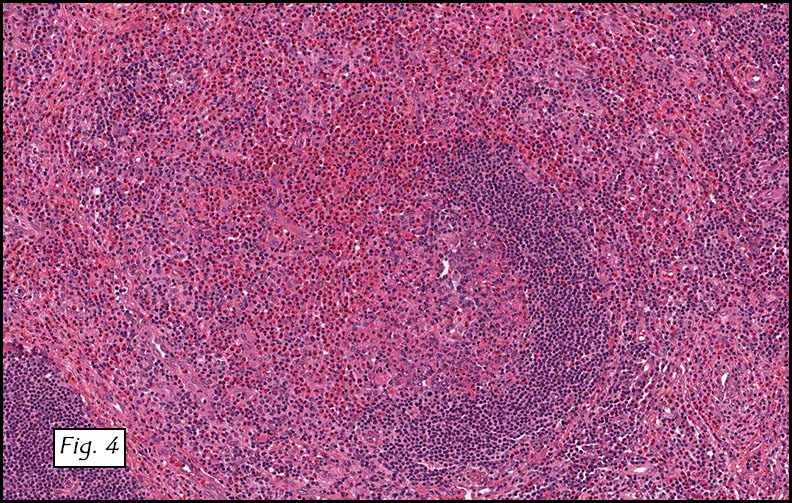
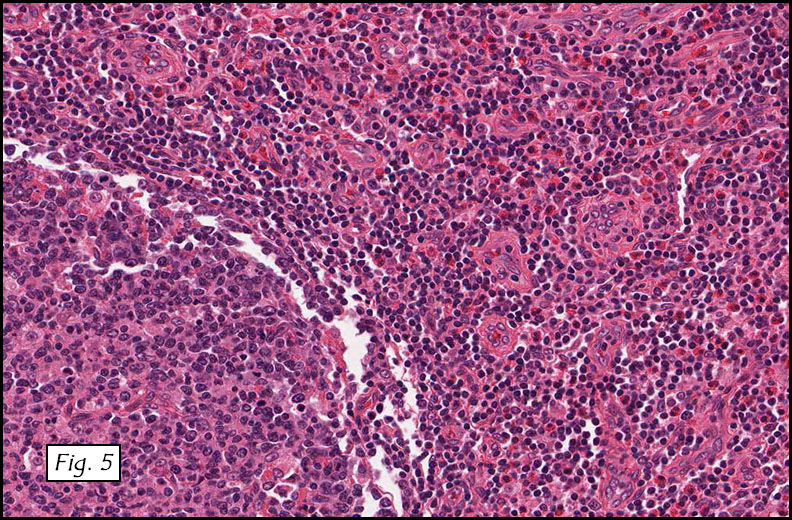
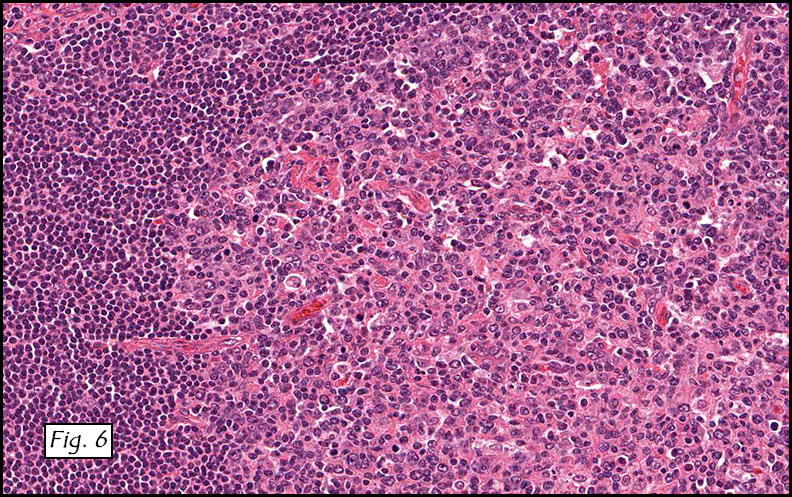
H&E staining of the central lesion revealed marked hyperplasia of lymphoid follicles with expanded germinal centers and well-formed mantle zones (Fig. 1). Some of the follicles had intervening fibrosis (Fig. 2). Germinal centers contained lymphocytes admixed with dendritic cells, polykaryocytes and eosinophilic proteinaceous deposits. Eosinophils were widely interspersed between the follicles with focal micro-abscess formation (Fig. 3), or occasionally infiltrated germinal centers with resultant follicular lysis (Fig. 4). The lesion was well-vascularized. Numerous post-capillary venules lined by flat endothelial cells were present between follicles and within germinal centers (Figs. 5, 6). Epithelioid endothelial cells were not seen.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Diagnosis: Kimura Disease, arm
Li Lei, M.D., Ph.D. and Donald R. Chase, M.D.
Department of Pathology and Human Anatomy
Loma Linda University Medical Center, Loma Linda, California
California Tumor Tissue Registry, Loma Linda, California
Discussion: Kimura disease (KD), named after Kimura for recognizing the vascular component of the entity in 1948, was first described in China in 1937 by Kim and Szeto as eosinophilic hyperplastic lymphogranuloma. It is a chronic inflammatory process characterized by benign reactive lymphoid proliferation with significant eosinophilia, secondary vascular proliferation and fibrosis.
KD is endemic in Asia, particularly in East and Southeast Asia. Other racial groups including Caucasian, African American and Hispanic may also be targeted. There is a striking male predominance with reported male-to-female ratio ranging from 3.5:1 to 19:1. Most of the affected patients are in their twenties to forties.
KD typically presents as a deep subcutaneous mass measuring 2 – 6 cm in size, but giant tumors up to 13 cm have been reported. Commonly it presents as cervical lymphadenopathy with a classic example being a peri-auricular mass involving the parotid gland. Less common sites include the extremities (like our case), axilla, groin, epiglottis, chest wall, spermatic cord and peripheral nerve.
Laboratory tests usually show peripheral blood eosinophilia and elevated serum immunoglobulin (IgE). Imaging is usually nonspecific, but sometimes may be seen as multiple ill-defined, enhancing lesions around the parotid gland with associated lymphadenopathy.
Clinically, up to 16% of cases are associated with proteinuria, nephrotic syndrome, membranous glomerulonephritis, bronchial asthma, ulcerative colitis or necrotizing eosinophilic vasculitis. Patients are otherwise asymptomatic.
Histologically, the tumor consists of dense lymphoid aggregates with prominent germinal centers, which contain IgE-bearing dendritic reticulum cells, polykaryocytes (Warthin-Finkeldey type), nuclear debris and a delicate eosinophilic matrix. Interfollicular dense eosinophilic infiltrate is a common feature and may form microabscesses. Plasma cells, small lymphocytes and mast cells are often increased in the paracortex. There is a moderate proliferation of postcapillary venules which are lined by flat endothelial cells. Long-standing lesions usually develop hyaline fibrosis and become less vascular. Affected lymph nodes demonstrate same histologic features with preserved architecture.
The differential diagnosis prominently includes “Epithelioid hemangioma†(EH), also known as “angiolymphoid hyperplasia with eosinophiliaâ€, a benign vascular neoplasm which shares many features of KD. Both entities affect young to middle-aged adults, presenting in the head and neck. Both have an inflammatory component, a vascular component and increased numbers of eosinophils, but there are differences:
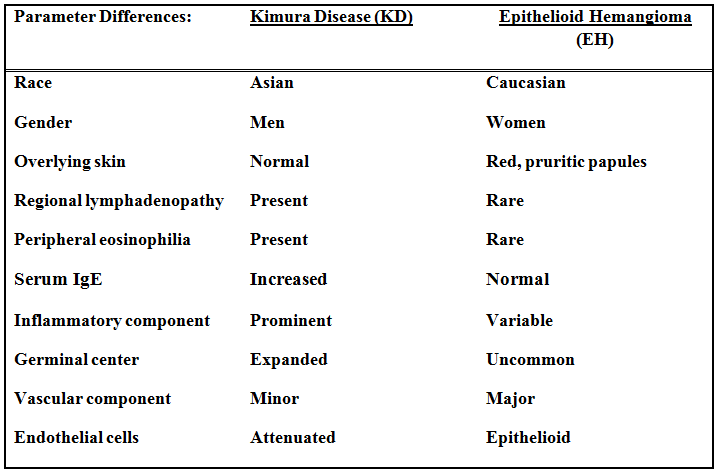
Despite the above differences (table) these two entities appear intertwined. Some lesions have characteristic features of both making specific classification almost impossible. Moreover, KD and EH can coexist in the same patient. Patients with one entity can develop the other during follow-up. These phenomena suggest that EH and KD may be a part of a spectrum of reactive vascular lymphoid proliferations.
In addition to EH, other differentials of KD include:
• Angioimmunoblastic T cell lymphoma at its early phase can have marked follicular hyperplasia with germinal centers, eosinophilia and prominent high endothelial venules. However, patients are often older and present with fever and generalized lymphadenopathy. Lymph node architecture is usually effaced. Perivascular clustering of neoplastic clear cells and increased follicular dendritic cell meshworks are helpful clues. Molecular study shows monoclonal T cell receptor gene rearrangements in the majority of the cases or IgH rearrangements in the minority.
• Langerhans cell histiocytosis can present as indolent lymphadenopathy in young adults. Microscopically, lymph node architecture is effaced by mixed infiltrates of Langerhans cells and eosinophils. CD1a and S100 immunoreactivity help to confirm the diagnosis.
• Follicular lymphoma: Primary cutaneous follicular lymphoma generally affects the elderly. Follicles consist of predominantly centrocytes with admixed centroblasts. In cases of secondary cutaneous involvement by follicular lymphoma, a pertinent medical history, back-to-back follicles with no cell polarization and t(14;18) translocation help reach the correct diagnosis.
• Hodgkin lymphoma has a bimodal age distribution with the first peak at 15-35 years of age. Lymph nodes have a “fish-flesh†cut surface. Despite lymphocytes, eosinophils, and collagen fibers in the background, CD30 and CD15 positive Hodgkin Reed-Sternberg cells are pathognomonic.
The etiology of KD remains unknown. Multiple clues such as elevated serum IgE, peripheral eosinophilia and association with other autoimmune diseases favor immune system hypersensitivity as the underlying pathophysiology. The endemic epidemiology of KD may reflect either inherent genetic susceptibility or an antigenic trigger unique to the geographic area.
Patients with KD normally have a favorable outcome. If untreated, the mass may remain stable or spontaneously regress. The standard care is simple surgical excision. Other options include laser therapy, radiotherapy and systemic corticosteroids. Local recurrence rate vary from 14 – 44%. Characteristics which may predict recurrence include: size ≥ 3.5 cm, peripheral eosinophilia ≥35%, Notch-1 upregulation and Ki-67 proliferation index ≥ 3% outside the germinal center. Metastases are very rare and therefore anecdotal.
Suggested Reading:
Goldblum J, Folpe A, Weiss S. Enzinger & Weiss’ Soft Tissue Tumors, 6th ed: Philadelphia, Elsevier Inc, 2014; 649-654.
Rosai J. Rosai and Ackerman’s Surgical Pathology, 10th ed: Philadelphia, Elsevier Inc, 2011; 1805.
Kung IT, Gibson JB, Bannatyne PM. Kimura’s disease: a clinico-pathological study of 21 cases and its distinction from angiolymphoid hyperplasia with eosinophilia. Pathology. 1984;16:39-44.
Deng WY, Ye SB, Luo RZ, Yan SM, Gao YF, Yang YZ, Guo ZM, Chen YF. Notch-1 and Ki-67 receptor as predictors for the recurrence and prognosis of Kimura’s disease. Int J Clin Exp Pathol. 2014;7: 2402-10.
Buder K, Ruppert S, Trautmann A, Bröcker EB, Goebeler M, Kerstan A. Angiolymphoid hyperplasia with eosinophilia and Kimura’s disease – a clinical and histopathological comparison. J Dtsch Dermatol Ges. 2014;12:224-8.
Liu XK, Ren J, Wang XH, Li XS, Zhang HP, Zeng K. Angiolymphoid hyperplasia with eosinophilia and Kimura’s disease coexisting in the same patient: evidence for a spectrum of disease. Australas J Dermatol. 2012;53:e47-50